前言
【为了好奇的研究:有些人为什么早睡早起?
以生物化学为发现途径,以遗传学为功能证据,研究生物钟。
我长期教课的一章是生物钟,但这是我实验室第一篇研究生物钟的研究论文】
我实验室的研究,多数是一两个人为主的,文章也就经常作者不多,甚至有时就一个研究生和我。我自己做研究生期间也是这样。但有时例外。今天简介的工作作者多,因为前赴后继用了很长时间,虽然每一个阶段是一个研究生为主。
2001年,旧金山加州大学(UCSF)的Fu和Ptacek实验室在分析人的时候,发现有一家人,凡是出现特定突变的,其昼夜节律(“生物钟)不是二十四小时,而且缩短,所以他们每晚7点睡觉、早上4点起床(“提前相位综合症”)。具体突变在Per2基因,改变它编码的一个氨基酸,让662位的S(丝氨酸)变成G(甘氨酸):S662G。
我们2012年开始找Per S662位点的激酶,到我们做完实验的2024年、发表文章的2026年距离发现现象的2001年是25年、距离我们开始这项工作是14年。
我们一开始没有特异抗体,那时读研究生的余腾辉试着用cDNA共表达筛选,找到磷酸化其他位点的激酶。然后通过北生所知道那个兔单抗的公司,一次就做成了抗S662磷酸化位点的特异抗体(在体外包括细胞可以,在体内不行),足以用于生化纯化的检测我们拿到CK1d和e,以及TSSK。
而在2018年,国外实验室发PNAS,他们发现(没有用生化提纯)CK1D和E磷酸化per 2 的S662。快而脏的研究(quick and dirty),但得到的结论一样(but made the point)。我们因此无法发表。
下面一个研究生李扬拼命做tssk的功能,但确实是它们却是睾丸特异的基因,不参与全身的生物钟,睾丸有没有生物钟?我们做不出来。失败了。
然后我们想救这文章,就在视上核(SCN)局部敲除 CK1,结果表型是生物钟减慢,与预期相反。
其他人后来也发表型,也是生物钟周期延长。这要是在那篇PNAS,就明显矛盾而无法发PNAS。
刘玉祥发现HEK细胞就有CK1之外的其他磷酸化PER2-662的蛋白激酶。
我们刘玉祥用生物化学分离纯化,找到磷酸化PER2-662位点的蛋白激酶MARKs。
它有四个类似的激酶(1-4)。一个一个MARK在D15培养细胞敲除,MARK2和3有生物钟表型而且是提前。好像MARK1没有、MARK4或者没有或者有
然后,在小鼠做基因剔除,MARK2肯定有生物钟表型,而且是提前,与S662突变的预计一样生物钟表型一样。
所以写文章了。
从2012年到2024年断断续续做实验,文章到2026才出来。
从好奇心上解决了困扰同领域科学家们的好奇。
但是,但只是部分,在这篇文章之后,可能还有工作。

摘要
遗传学一直是研究生物钟的重要方法,揭示了第一个生物钟的关键调控基因Period(Per)。2001年发现人类PER2基因第662 位点的丝氨酸突变成甘氨酸(S662G)会导致家族性睡眠状态提前综合征(FASP),表现为活动起始时间提前和生物钟周期缩短。本研究发现,PER2 S662被CK1δ/ε、TSSK1/2及SIK1-3磷酸化,但分别敲除上述7个基因后均未观察到活动起始时间提前的表型。进一步通过生化纯化实验发现,MARK2磷酸化S662位点,结合并稳定PER2蛋白。在Mark2缺陷型细胞中,细胞节律周期以S662位点依赖性的方式缩短。神经元特异性敲除Mark2基因的小鼠表现出活动起始时间提前和周期缩短。本研究揭示了MARK2作为生物钟重要调控因子的生理功能,同时证实了生化纯化方法在行为机制研究中的有效性。
关键词
生物钟,PER2,S662磷酸化,FASP,生化纯化,MARK2
研究意义
生物钟调节生理和行为的日常节律,其紊乱与睡眠障碍等多种健康问题密切相关。PERIOD2 (PER2) 是生物钟的核心调控因子。2001年研究发现,人类PER2基因中第662位丝氨酸残基突变为甘氨酸(S662G)会引起家族性睡眠状态提前综合征(FASP),表现为活动起始时间提前和周期缩短。该突变消除了S662的磷酸化修饰,而S662的磷酸化通常作为分子开关,稳定PER2并延长生物钟周期。过去25年间,寻找并鉴定磷酸化S662的激酶一直是该领域的研究热点。本研究通过生化纯化技术,首次确定MARK2(一种AMPK相关激酶家族成员)是PER2 S662的新激酶。对所有21种AMPK相关激酶的进一步检测发现,其中部分成员可磷酸化PER2 S662。MARK2能够直接磷酸化PER2,与之结合,并通过S662磷酸化延缓其降解。在细胞中敲除MARK2基因导致细胞节律缩短,且该表型依赖于PER2 S662的磷酸化。神经元特异性敲除Mark2基因的小鼠表现出活动起始时间提前和周期缩短。本研究不仅证明了MARK2参与调控生物钟,也强调了生化方法在解决生理学难题中的有效性,并提示AMPK相关激酶可能通过调节PER2及其他蛋白的磷酸化参与生物钟调控。
引言
生物钟对基本生理功能至关重要,其异常与健康状况密切相关。每年有超过十亿人次的跨时区旅行者和约20%的西方工作者从事轮班工作¹,与生物钟紊乱相关的健康问题日益受到关注⟡⁻⁷。
生物钟是一个内在的计时系统,驱动着睡眠/觉醒周期、免疫反应和新陈代谢等多个系统的日常节律⁸⁻¹⁰。在哺乳动物中,主时钟位于下丘脑的视交叉上核(SCN),负责协调全身细胞的生物钟振荡¹¹‚¹⟡。50余年前,借助遗传学方法在果蝇¹³、脉孢菌¹⁴和衣藻¹⁵中发现影响生物钟的基因突变,使人类对生物钟分子机制的理解取得了重大突破。此后,在多种生物(尤其是果蝇和小鼠)中的遗传学研究,确立了相互连锁的转录-翻译反馈环(TTFL)作为生物钟的分子机制¹⁶⁻⟡⁴。
第一个被发现从果蝇到人类均保守的关键基因是Period(Per)¹³。哺乳动物中存在两个转录-翻译反馈环:在第一个TTFL中,转录因子BMAL1和CLOCK形成异源二聚体,激活3个Per基因和2个Cry基因的转录;随后,翻译产生的PER蛋白与其他蛋白形成大分子复合物,反馈抑制自身转录¹⁹‚⟡⁵⁻³⁶。第二个TTFL涉及BMAL1和CLOCK激活两个核受体基因(Rorα/β,Rev-erbα),而翻译产生的RORα/β和REV-ERBα蛋白则反馈调节Bmal1的转录³⁷。近期,张二荃实验室发现核苷三磷酸酶(NTPase)也是保守的生物钟调控因子³⁸。
在核心生物钟调控蛋白中,PER家族成员在蛋白丰度上表现出最显著的节律振荡³⁹⁻⁴¹。合成后,PER2蛋白经历多位点逐步磷酸化,以调节其稳定性、抑制子活性和亚细胞定位³³‚³⁴‚³⁶‚⁴¹⁻⁴⁶。有两个磷酸化区域对PER2蛋白稳定至关重要:S478/482 phosphodegron(小鼠),其磷酸化后招募E3泛素连接酶β-TrCP,促进PER2降解,从而加速生物钟周期;FASP区域(从小鼠S659或人S662起始),由五个丝氨酸组成(SxxSxxSxxSxxS),其磷酸化可抑制phosphodegron的磷酸化,从而稳定PER2并延长生物钟周期³³‚⁴¹⁻⁴⁴‚⁴⁷⁻⁵⁶。S662的磷酸化是FASP区域内后续磷酸化的先导事件⁴¹‚⁵¹‚⁵⁷⁻⁵⁹。人类S662G突变消除了这一先导磷酸化,阻断了后续FASP位点的磷酸化⁴¹‚⁴⁸‚⁵¹‚⁶⁰,导致家族性睡眠状态提前综合征,生物钟周期缩短约4小时。后续研究在转基因小鼠中成功重现了这一表型⁴³‚⁴⁸。
2001年的一项研究通过对一个家族的遗传学分析,发现人类PER2蛋白(hPER2)第662位氨基酸由丝氨酸(Ser)突变为甘氨酸(Gly)与家族性睡眠状态提前综合征(FASPS)相关⁴³‚⁶¹。hPER2 S662是酪蛋白激酶1δ (CK1δ)的先导磷酸化位点⁴³‚⁵¹‚⁵⁷‚⁵⁸。CK1δ和CK1ε被认为是PER2磷酸化的关键激酶,至少磷酸化小鼠PER2三个区域:phosphodegron(S478)、FASP先导磷酸化位点(S659)及FASP区域内的下游丝氨酸³³‚⁵⁰‚⁵¹‚⁶⟡‚⁶³。CK1δ/ε与PER2形成稳定复合物,通过其激活环的构象变化及C末端尾部磷酸化状态的改变来切换底物偏好,使PER2蛋白稳定性呈现开关样变化³³‚³⁴‚³⁶‚⁵⁶⁻⁵⁹‚⁶⁴。然而,在HEK293细胞中同时敲除CK1δ和CK1ε后,S662磷酸化水平显著降低但未完全消除,提示存在其他作用于该位点的激酶。
在长达15年寻找PER2 S662上游激酶的过程中,我们尝试了分子生物学和生物化学两种方法。在获得特异性识别hPER2磷酸化S662的抗体之前,我们将人类激酶组中的蛋白激酶cDNA与hPER2共转染,发现了几种能够磷酸化hPER2片段的激酶。在获得PER2 S662磷酸化单克隆抗体(mAb) 后,我们确认了CK1 δ和ε、TSSK1和2磷酸化PER2 S662位点。在所有CK1中,只有CK1δ和ε,而非CK1γ1、2或3,能在体外磷酸化S662。在D15细胞系中敲除CK1 δ和ε后,细胞节律周期延迟。第二种策略是使用抗磷酸化S662-PER2抗体进行生化纯化,这使我们发现了MARK2、3是PER2 S662的激酶。它们属于AMPK相关激酶家族(ARK) 的成员⁶⁵-⁷⁷。我们比较了所有ARK激酶家族的体外活性,发现SIK 1-3、MARK 1-4、TSSK1和2磷酸化PER2 S662位点,其中MARK2和MARK3活性最强。敲除SIKs 未显示活动起始时间提前的表型⁷⁸-⁸⁰。TSSK1和2敲除小鼠(包括它们的双敲除),也未发现活动起始时间提前的表型。因此,CK1s、SIKs和TSSKs敲除小鼠的生物钟表型与人类FASPS患者⁴⁸ 或 PER2 S662G 突变小鼠⁴³ 在体内的表型不一致。
在D15细胞中过表达MARK2或3延长了细胞节律,敲除MARK2(而非MARK3)则缩短了节律,这与S662G突变的表型相似,进一步生化和遗传学证据证明MARK2在分子和生理上都位于PER2 S662的上游。在小鼠神经元敲除MARK2 导致活动起始时间提前和生物钟缩短,而MARK3敲除、MARK3神经元特异性敲除和MARK4神经元特异性敲除小鼠未表现出生物钟异常。因此,我们长期寻找PER2 S662激酶的努力揭示了MARK2是一个在生物钟调控中具有生理意义的激酶。
结果
CK1 δ和ε磷酸化hPER2 S662与生物钟表型
早期研究曾报道CK1δ/ε可磷酸化PER2⁴¹,⁴⁸,⁶⟡,⁸¹,2005年被认为是PER2 S662的激酶⁴⁶,同一研究团队因未能在体外反应体系中检测到PER2 19氨基酸残基肽段中S662的直接磷酸化,推翻此观点⁴³。这一争议直至2018年才由Narasimamurthy等人解决,他们证实了CK1δ/ε可直接磷酸化PER2 S662位点⁵¹。近年结构和生化研究阐明了其调控PER2磷酸化的分子机制:CK1激活环的构象转换控制其对PER2的底物选择性,进而调控PER2的磷酸化位点,调控PER2稳定性开关;PER2 FASP区域的磷酸化可别构抑制CK1活性,确保生物钟周期的精确控制⁵³‚⁵⁴‚⁵⁶⁻⁵⁹。
在获得识别PER2 662位磷酸化的特异性抗体前,我们采用Phos-tag技术,通过SDS-PAGE检测磷酸化引起的凝胶迁移⁸⟡,筛选了包含288种人源激酶的cDNA文库(表S1),检测其对6个hPER2片段的磷酸化能力。将激酶cDNA与hPER2片段在HEK293T细胞中共转染后,发现数种可磷酸化特定PER2区域的激酶:PER2 1-200未被任何筛选激酶磷酸化;CDK5磷酸化PER2 150-400;PRKACB、PRKG1和IKKβ磷酸化PER2 328-556;CK1δ/ε、TSSK2和IKKε磷酸化PER2 556-771。为明确CK1δ/ε是否磷酸化hPER2 S662,我们获得了特异性识别磷酸化S662 hPER2的单克隆抗体。该抗体特异性识别磷酸化的野生型人/小鼠PER2,但不识别S662A/S662D(hPER2)或S659A/S659D(mPER2)突变体(图S1A-C)。磷酸酶处理可消除其识别信号(图S1A-C)。
利用此抗体,我们证实了CK1δ/ε在细胞中磷酸化hPER2 S662位点(图S1D-H)。FLAG标记的CK1δ(图S1D)或CK1ε(图S1E)在HEK293细胞中磷酸化hPER2 556-771片段的S662。梯度转染CK1δ或CK1ε质粒可增加hPER2 S662位点的磷酸化水平(图S1G)。对其他CK1家族成员(CK1α1,α2,γ1,γ2,γ3)的检测显示,无论是将其共转染至HEK细胞(图S1F),还是采用免疫沉淀的CK1蛋白与重组hPER2片段进行体外激酶测定(图S1H),均未发现这些CK1可磷酸化PER2 S662。
鉴于全长CK1δ/ε的C末端自磷酸化可导致自身抑制,我们在大肠杆菌中表达了其截短变体(1-317)的重组蛋白,以确认其直接磷酸化作用。重组CK1δ 1-317(图S2 A,B,E,G)和CK1ε 1-317(图S2 C,D,F,G)可磷酸化细菌表达的hPER2 556-771片段上的S662位点,而其他CK1则无此活性(图S2 G,J)。CK1δ作用于PER2 S662 321-771的米氏常数(Km)为6.58±1.71 μM(图S2H),CK1ε的Km为7.38±1.61 μM(图S2I)。
为探究CK1δ/ε在细胞节律中的功能,我们首先采用Per2-dLuc细胞(D15细胞),一种改造的人U2OS细胞系,携带由Per2启动子驱动表达的荧光素酶报告基因(Per2::luciferase),可用于监测细胞节律(图S3C)⁸³。在D15细胞中敲除CK1δ(图S3A)或CK1ε(图S3B)显著延长了细胞节律周期(图S3 C,D),与既往报道一致³⁵。进一步利用基因敲除小鼠研究CK1δ的生理功能,发现神经元条件性敲除CK1δ小鼠的生物钟周期延长约0.3小时(图S3 E,F)。本研究结果(图S1-3)与文献报道一致:尽管CK1δ/ε可在体外磷酸化PER2 S662,但在细胞系或小鼠中敲除CK1δ/ε均导致周期延长³⁵‚³⁶‚⁴¹‚⁴³‚⁵⁰‚⁵⁴‚⁵⁷‚⁸⁴⁻⁸⁹。更重要的是,在HEK293细胞中同时敲除CK1δ/ε后(图S3 A,B),S662磷酸化信号虽降低但未完全消除(图S3 G,H),提示存在作用于该位点的其他激酶。
TSSK1/2磷酸化hPER2 S662与生物钟表型
初期cDNA筛选发现TSSK2也是PER2 556-771片段的激酶。我们也将编码TSSK家族激酶的cDNA与hPER2 556-771在HEK293T细胞中共转染,结果显示TSSK1和TSSK2(而非TSSK3/4/6)可增强PER2 S662磷酸化(图S4A)。在HEK细胞中增加TSSK1/2 cDNA的浓度,提高了PER2 S662磷酸化水平(图S4B)。
为确定Tssks在生物钟调控中的生理作用,我们构建了Tssk2基因敲除小鼠(图S4C)及Tssk1-Tssk2条件性双敲除小鼠(图S4D)。结果显示,无论是Tssk2单敲除(图S4 E-H)还是神经元特异性双敲除Tssk1& Tssk2小鼠(图S4 I-L),均未表现出任何生物钟表型。
通过生化纯化发现MARK2/3为PER2 S662激酶
鉴于CK1δ/ε双敲除细胞中仍残留部分S662磷酸化,我们采用抗磷酸化S662单克隆抗体监测活性,进行了激酶的生化纯化(图1A)。
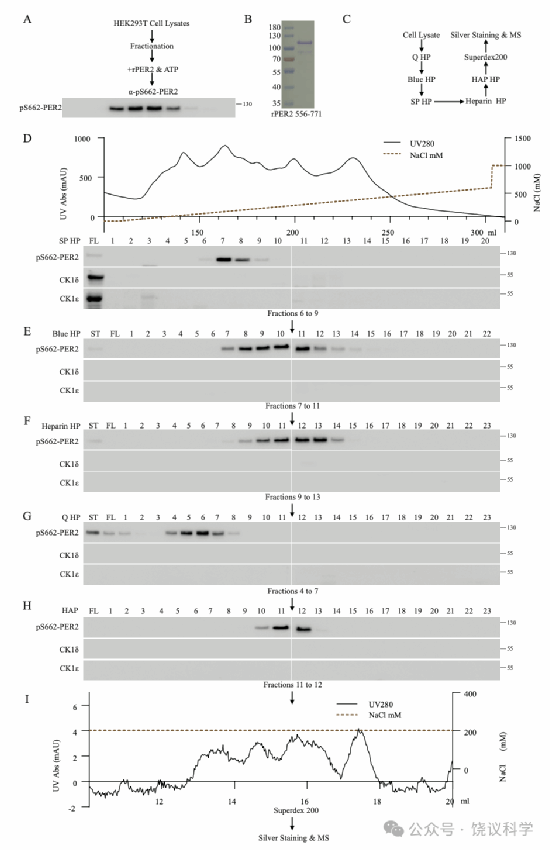
图1.从HEK293细胞中纯化磷酸化PER2 S662的活性组分
(A)检测HEK293细胞提取物各组分中磷酸化PER2 S662的活性组分的实验流程示意图。将各分离纯化的组分与1 μg重组PER2 556-771(rPER2 556-771)及1 mM ATP在37°C孵育1小时,随后采用磷酸化S662的特异性抗体进行免疫印迹检测。(B)从大肠杆菌中重组表达的MBP-hPER2 556-771-GFP-8His底物的考马斯亮蓝染色结果。(C)PER2 S662激酶的六步层析纯化策略。以约50升悬浮培养的HEK细胞为起始材料,正式纯化前先采用相当于起始材料10%的样品在六个色谱柱上优化纯化方案。(D)SP HP阳离子交换层析柱分离纯化各组分中磷酸化PER2 S662的活性组分检测。在组分7和8(210-240 mM NaCl)中检测到磷酸化PER2 S662的活性组分,而CK1δ和CK1ε仅存在于流穿组分(0 mM NaCl)中。(E-I)将SP HP柱的活性组分(组分7和8)依次通过后续五个色谱步骤进行纯化,顺序为Blue HP亲和层析柱(E)、Heparin HP亲和层析柱(F)、Q HP阴离子交换层析柱(G)、HAP羟基磷灰石层析柱(H)和Superdex 200凝胶过滤层析柱(I)。合并每个层析柱的活性组分并上样至下一层析柱。相关数据参见附图S1-4及附表S1。
以50升悬浮培养的HEK293T细胞(2.0-3×10⁶细胞/mL)提取物为起始材料(图1A),以从大肠杆菌纯化的重组hPER2 556-771为底物(图1B)。在全面纯化前,先用相当于起始材料10%的量,在六个色谱柱(SP HP,Blue HP,Heparin HP,Q HP,HAP和Superdex 200)上测试了纯化方案(图1C)。每步纯化均监测各洗脱组分中hPER2-S662的磷酸化活性。将含500 mg蛋白的裂解液(浓度10 mg/mL)上样至10 mL SP HP柱,以NaCl梯度洗脱,在组分7和8(210-240 mM NaCl)中检测到强PER2 S662磷酸化活性,而CK1δ/ε仅存在于流穿组分(0 mM NaCl)(图1D)。合并活性组分依次上样:Blue柱组分7-11(1050-1650 mM NaCl)(图1E),Heparin柱组分9-13(270-330 mM NaCl)(图1F),Q HP柱组分4-7(120-210 mM NaCl)(图1G),HAP柱组分11-12(150-180 mM K₂PO₄)(图1H)。将HAP柱活性组分经Superdex 200柱分离,以200 mM NaCl洗脱(图1I)。
将Superdex 200柱组分透析后进行SDS-PAGE及银染(图2A)。从银染凝胶的组分11中切取所有条带进行质谱分析,检测到6种蛋白激酶:MARK3、MARK2、GSK-3β、MKNK1、SRC和RSK2(图2B)。
MARK激酶磷酸化PER2 S662位点
在HEK细胞中表达上述激酶的N端FLAG标签的融合蛋白(图2C)⁹⁰,免疫沉淀各FLAG标记激酶后,检测其对大肠杆菌来源重组hPER2 556-771的磷酸化能力。结果显示GSK-3β、MKNK1、SRC和RSK2不能磷酸化hPER2-S662,而MARK2/3可强烈磷酸化hPER2-S662(图2C)。将PER2 S662突变为A后,MARK2(图2D)和MARK3(图S5A)均无法磷酸化该位点。
MARK2/3通过其activation loop中保守的苏氨酸残基(MARK2 T208,MARK3 T211)磷酸化而被激活。将T208(MARK2)(图2D)或T211(MARK3)(图S5A)突变为A后,突变激酶无法磷酸化PER2-S662。将MARK2 T208突变为谷氨酸(E)(图2D)或MARK3 T211E(图S5A)降低了其磷酸化能力。在HEK细胞中转染递增浓度的野生型或T208E突变型MARK2 cDNA(图2E),或野生型或T211E突变型MARK3 cDNA(图S5B),均可增强hPER2-S662的磷酸化水平。小鼠来源的MARK2/3同样可磷酸化hPER2-S662(图S5 C,D)。
MARK家族包含四个成员(MARK1-4)。在HEK细胞中过表达任一MARK成员均增强PER2 S662磷酸化(图2 F,G)。在CK1δ/ε双敲除HEK293细胞中分别敲除MARK1-4,PER2 S662磷酸化分别减少21%、43%、50%和47%(图2 H,I)。在HEK293细胞中同时敲除所有四个MARK基因后(图S5 E-H),三个独立敲除细胞系中hPER2-S662的磷酸化水平显著降低60%-67%(图S5 I,J)。

图2. MARK2和MARK3磷酸化PER2 S662
(A)Superdex 200凝胶过滤层析柱各组分银染结果及磷酸化PER2 S662的组分活性测定。在组分11中检测到磷酸化PER2 S662的活性组分,将该组分经切胶后进行质谱分析。(B)质谱鉴定出的六种激酶,按Mascot分数从高至低排列。其中SRC为酪氨酸蛋白激酶,其余均为丝氨酸/苏氨酸蛋白激酶。(C)hPER2 S662可被免疫沉淀的MARK2和MARK3在体外磷酸化,而GSK3β、MKNK1、SRC及RSK2无此活性。将FLAG标签的激酶在HEK293细胞中表达,免疫沉淀后与重组hPER2(556-771)底物进行体外激酶反应。(D)rPER2 S662在体外可被从HEK细胞免疫沉淀的野生型MARK2及其T208E活化突变体磷酸化,而T208A失活突变体无此活性。(E)野生型MARK2及其T208E突变体以剂量依赖性方式增加PER2 S662磷酸化水平。将递增浓度的MARK2 cDNA转染HEK293T细胞,培养24小时后收取细胞裂解液进行免疫印迹分析。(F)在HEK细胞中过表达各MARK家族成员(MARK1-4)均可增强PER2 S662磷酸化水平。(G)采用ImageJ软件对(F)进行定量分析(n=3个独立生物学重复)。(H)在CK1δ/ε双敲除HEK293细胞中分别敲除各MARK基因后,PER2 S662磷酸化水平均出现降低。(I)采用ImageJ软件对(H)进行定量分析(n=3个独立生物学重复)。统计分析采用单因素方差分析-Dunnett多重比较检验(G和I);数据以均值±标准误表示。相关数据参见附图S5。
MARK2/3直接磷酸化PER2的生化特性
前述实验均在HEK细胞中进行。为探究MARK2/3是否直接磷酸化hPER2 S662,我们在大肠杆菌中表达了这些激酶的重组形式。结果显示,T208E突变型MARK2磷酸化hPER2 S662(而非PER2 S662A或S662D),而野生型或T208A突变型MARK2无此活性(图3A,B)。MARK3 T211E突变体呈现相似结果(图S6A,B)。并且MARK2对PER2片段的Km值(6.54 ± 2.06 µM)(图3C)显著低于MARK3(23.04 ± 5.99 µM)(图S6C)。这一差异可通过后续发现解释:MARK2与PER2 498-556区域存在特异性结合,而MARK3无此结合(图5A,5D)。此外,重组小鼠MARK2 T208E(图3D)和MARK3 T211E(图S6D)磷酸化mPER2 S659,但对S659A或S659D突变体无活性。综上,重组的MARK2 T208E和MARK3 T211E的突变体可直接磷酸化PER2 S662位点。
观察发现MARK2 T208E和MARK3 T211E突变体具有活性而野生型无活性,是由于野生型需在哺乳动物细胞中磷酸化激活,而大肠杆菌表达系统无法提供此修饰。MARKs属于AMPK相关激酶家族,其成员可被LKB1或STE20家族成员激活⁷⁷‚⁹¹。因此,我们检测了STE20亚家族激酶MAP4K7的作用。用重组MAP4K7处理从大肠杆菌纯化的野生型MARK2/3后,二者激活环发生磷酸化(图3E)。经MAP4K7预处理的野生MARK2/3能够磷酸化PER2 S662(图3E)。类似地,MAP4K7预处理可激活从大肠杆菌纯化的野生重组小鼠MARK2/3,使其能够磷酸化mPER2 S659(图3F)。这些结果表明,MARKs是直接的PER2 S662激酶,可被STE20亚家族等上游激酶激活⁷⁷‚⁹¹,提示研究MARKs在调控机制具有重要意义。

图3.重组MARK2体外直接磷酸化PER2 S662本图所有实验均使用在大肠杆菌中表达的重组蛋白进行反应。hPER2底物(rPER2野生型、S662A、S662D突变体)及小鼠PER2底物(r-mPER2野生型、S659A、S659D突变体)均包含556-771位氨基酸残基。rMARK2为在大肠杆菌中表达的重组人源MARK2蛋白。rMAP4K7为重组人源MAP4K7(1-500位氨基酸)蛋白。
(A-B)重组MARK2 T208E活化突变体直接磷酸化hPER2的S662位点,而野生型MARK2及T208A失活突变体无此活性;MARK2 T208E对S662A及S662D突变体底物均无磷酸化作用。(C)重组MARK2 T208E作用于PER2的米氏常数测定。将约0.2 μg rMARK2 T208E与不同浓度的重组rPER2 321-771底物于37°C孵育30分钟。免疫印迹结果采用ImageJ软件进行定量分析(n=3个独立生物学重复),Km值采用GraphPad软件计算。数据以均值±标准误表示。(D)重组小鼠MARK2 T208E磷酸化小鼠PER2的S659位点。(E)经rMAP4K7预激活的重组野生型MARK2及MARK3能够磷酸化重组hPER2 S662。MAP4K7为MARKs的上游激酶。将从大肠杆菌纯化的重组MARKs与MAP4K7预孵育1小时后,检测其对重组hPER2的激酶活性。采用抗磷酸化MARK2/3抗体检测MARK激活环的磷酸化水平,以确认MARK2/3的活化状态。(F)MAP4K7预处理可激活重组小鼠MARK2及MARK3,使其获得磷酸化mPER2 S659的能力。相关数据参见附图S6。
AMPK相关激酶家族成员对hPER2 S662的体外磷酸化
MARKs和TSSKs是AMPK相关激酶家族成员⁶⁵⁻⁷⟡,我们前2篇文章对该家族进行了系统研究⁷⁷‚⁹¹。SIK1/3亦属此家族,是已知的生物钟调控蛋白:在视交叉上核中敲低Sik1可导致节律快速时相偏移,增强对时差反应的抵抗力⁹⟡;SIK3则促进PER2去稳定化,其敲除可延长生物钟周期⁷⁸。
本研究检测了所有AMPK相关激酶对hPER2 S662的活性(图4),包括从HEK细胞免疫沉淀的ARKs(图4 A-D,J)和从大肠杆菌纯化的ARKs(图4 E-H,J;图S7-9)。从HEK细胞免疫沉淀的ARKs为FLAG标记的全长野生型蛋白(图4A-D,J);从大肠杆菌纯化的ARKs则为全长或含激酶结构域的截短蛋白,有野生型或激活环磷酸化位点突变(T→E)两种形式(图4E-H,J;图S7)。从大肠杆菌纯化的野生型ARKs在检测对hPER2 S662的激酶活性前,均经MAP4K7预处理(图4 G-H)。结果显示,SIK1-3、MARK1-4、TSSK1/2/4及NIM1K可磷酸化PER2-S662(图4 A-J),其中MARK2、TSSK4和MARK3活性最强。TSSK4仅在从HEK细胞免疫沉淀时可磷酸化PER2-S662,从大肠杆菌纯化后则无此活性(图4J)。值得注意的是,免疫沉淀的全长CK1δ/ε活性中等,而去除自抑制C末端尾部的截短型CK1δ/ε (1-317)则具有高活性(图4C,I)。
无论从HEK细胞免疫沉淀或大肠杆菌纯化,BRSK1/2、AMPK1/2、NUAK1/2、MELK、SNRK、TSSK3/6均不能磷酸化PER2 S662(图4J)。TSSK4仅在从HEK细胞免疫沉淀时(而非大肠杆菌纯化时)磷酸化PER2 S662(图4J)。

图4. AMPK相关激酶家族对PER2 S662的磷酸化作用
(A-D)在HEK293细胞中分别表达FLAG标签的全长AMPK相关激酶及CK1相关激酶,免疫沉淀后检测其对重组hPER2 556-771(rPER2)的激酶活性。所有检测中均以SIK3作为信号强度参照。(E-H)从大肠杆菌分别纯化FLAG标签的全长或截短型AMPK相关激酶及CK1相关激酶,检测其对重组PER2 556-771的激酶活性。(E和F)对于野生型ARK无法磷酸化rPER2的情况,从大肠杆菌纯化其激活环T位点突变为E的活化型突变体(附图S7),检测其对rPER2的活性。从大肠杆菌纯化的野生型ARK重组蛋白,在检测其对重组hPER2的活性前,先与MAP4K7预孵育1小时。(I)从大肠杆菌分别纯化FLAG标签的CK1相关激酶,检测其对rPER2的活性。重组CK1δ和CK1ε为包含激酶结构域(1-317位氨基酸)的N端截短蛋白,其余CK1家族成员为全长蛋白。(J)ARKs对PER2 S662磷酸化能力的系统发育树总结。“IP”列表示从HEK293细胞免疫沉淀的22种激酶磷酸化PER2 S662的能力;“WT”和“E”列分别表示在大肠杆菌中表达的22种野生型激酶及其激活环磷酸化T位点突变为E的突变体形式磷酸化PER2 S662的能力。相关数据参见附图S7-9。
MARK2以S662依赖性方式结合并稳定PER2
随后采用抗FLAG抗体磁珠进行免疫共沉淀,检测MARKs与hPER2的相互作用(图5)。结果显示,MARK2(而非MARK3)与全长PER2特异性相互作用(图5A),并通过FLAG-MARK2与HA-PER2的反向免疫共沉淀进一步得到证实(图5B)。
为确定PER2中与MARK2相互作用的结构域,将hPER2分为四个片段(1-438,321-557,477-788,717-1255)。MARK2与全长PER2及片段321-557和477-788相互作用,但不与PER2 1-438或717-1255相互作用。进一步定位显示,MARK2与PER2 498-557区域特异性结合(图5D)。该相互作用结构域与S662磷酸化位点不同,提示MARK2可能先通过与PER2结合进而磷酸化S662位点。
已知PER2 S662磷酸化可增强其蛋白稳定性,而S662G突变则导致不稳定⁴¹‚⁴³‚⁴⁶‚⁴⁸‚⁵¹。为探究MARK2是否调节PER2稳定性,将PER2(野生型或S662G突变体)单独或与MARK2共转染HEK细胞,采用放线菌酮阻断蛋白翻译后检测PER2稳定性。结果显示,共转染MARK2可增强PER2稳定性(图5 E,F),表明MARK2参与PER2稳定性调节。然而,MARK2转染对PER2 S662G突变体稳定性无影响(图5 G,H),表明MARK2对PER2稳定性的调节依赖于S662位磷酸化。

图5. MARK2与PER2蛋白的相互作用及其对PER2稳定性的调控
(A-B)MARK2与PER2存在特异性相互作用。将FLAG标签的MARK2与HA标签的全长PER2共转染HEK293细胞,采用抗FLAG磁珠进行免疫共沉淀(A)。同时采用FLAG-MARK2与HA-PER2进行反向免疫共沉淀验证(B)。(C-D)MARK2与PER2的498-557区域特异性结合。将FLAG标签的MARK2与HA标签的各PER2截短片段及全长PER2共转染HEK293细胞,采用抗FLAG磁珠进行免疫共沉淀。从PER2 321-717片段中删除498-557位氨基酸可完全消除PER2与MARK2的相互作用。(E-H)MARK2以S662磷酸化依赖性方式延缓PER2蛋白降解。将PER2野生型(E)或S662G突变体(G)单独转染或与MARK2共转染HEK293细胞24小时。转染后细胞经200 μM放线菌酮处理,在指定时间点收集蛋白样品。免疫印迹结果采用ImageJ软件进行定量分析(n=3个独立生物学重复),数据采用GraphPad软件进行统计分析(F和H)。MARK2共表达可减缓野生型PER2的降解速率(E和F)。PER2 S662G突变体的降解速率快于野生型PER2,且与MARK2共表达对其降解速率无显著影响(G和H)。统计分析采用双因素方差分析-Sidak多重比较检验(F和H),数据以均值±标准误表示。
MARK2以S662依赖性方式调控细胞节律
为探究MARKs在生物钟调节中的作用,首先在D15细胞中分别过表达四个MARK成员。结果显示,过表达任一MARK成员均显著延长D15的细胞节律周期(图6A为MARK2/3,图S10 F-H为MARK1/4)。
为确定D15细胞节律调节所必需的Mark基因,分别敲除各基因后发现,Mark3(图6C)或Mark1(图S10 I,K)敲除细胞无节律变化;Mark4敲除细胞表现出轻微的周期缩短(图S10 J,K);Mark2敲除细胞周期显著缩短约1.13小时(图6C)。
为确定MARK2是否以S662依赖性方式调节细胞节律周期,将PER2 S662G突变引入D15细胞(图S10L)。与野生型细胞相比,S662G突变细胞节律周期显著缩短(图6E)。此外,虽然MARK2敲除在野生型细胞中缩短周期(图6C),但在PER2 S662G突变细胞中未能缩短周期(图6G),表明MARK2以S662依赖性方式调节细胞节律周期。
生化分析结果与此一致:过表达MARK2/3可增强PER2 S662磷酸化并提高PER2蛋白稳定性(图6B);而MARK2敲除则同时降低二者(图6D)。PER2 S662G突变细胞的PER2水平低于野生型细胞(图6F)。重要的是,MARK2敲除可降低PER2野生型细胞中PER2蛋白水平(图6D),但此效应在PER2 S662G突变细胞中完全消失(图6H)。综上所述,MARK2是PER2 S662上游激酶,以S662依赖性方式调节PER2稳定性及细胞节律长度。

图6. MARK2通过PER2 S662调控细胞节律周期
(A)过表达MARK2或MARK3分别使D15细胞节律周期延长0.63小时或1.63小时。采用慢病毒系统分别构建稳定过表达MARK2或MARK3的D15细胞系。展示代表性振荡曲线及周期统计结果(对照组:n=7;MARK2过表达组:n=8;MARK3过表达组:n=8个独立生物学重复)。(B)过表达MARK2或MARK3增加PER2 S662磷酸化水平及总PER2蛋白水平。过表达MARK2或MARK3对ERK、JNK及GSK3β的磷酸化水平及总蛋白水平均无显著影响。免疫印迹结果采用ImageJ软件进行定量分析(n=3个独立生物学重复)。(C)敲除MARK2(而非MARK3)显著缩短D15细胞的节律周期(对照组:n=7;MARK2敲除组:n=6;MARK3敲除组:n=6个独立生物学重复)。(D)MARK2敲除降低PER2 S662磷酸化水平及总PER2蛋白水平,但对ERK、JNK及GSK3β无显著影响。免疫印迹结果采用ImageJ软件进行定量分析(n=3个独立生物学重复)。(E-F)PER2 S662G突变使D15细胞节律周期缩短至20.01小时(对照组:n=6;S662G突变组:n=7个独立生物学重复),并降低PER2蛋白丰度。通过PCR及测序确认PER2 S662G突变成功引入D15细胞(附图S10L)。免疫印迹结果采用ImageJ软件进行定量分析(n=3个独立重复)。(G-H)在S662G突变细胞中,MARK2敲除无法进一步缩短节律周期(S662G组:n=8;S662G & MARK2双敲除组:n=8个独立重复)或降低PER2蛋白丰度(n=3个独立重复)。免疫印迹结果采用ImageJ软件进行定量分析(n=3个独立重复)。统计分析采用单因素方差分析-Dunnett多重比较检验(A-D);非配对Student's t检验(E-H)。数据以均值±标准误表示。相关数据参见附图S10及附表S3。
Mark2(而非Mark3)参与小鼠生物钟调节
为探究MARK激酶在生物钟调节中的生理功能,我们构建了Mark2和Mark3基因敲除小鼠(图S11)。由于Mark2敲除小鼠胚胎致死,我们构建了Mark2条件性敲除小鼠(图S11A),并与Nestin-Cre小鼠杂交,实现中枢神经系统神经元特异性Mark2敲除。小鼠首先在12小时光照/12小时黑暗周期中记录运动活动14天,随后在持续黑暗条件下记录运动活动21天(图7)。
Nestin-Cre; Mark2flox/flox小鼠的生物钟与对照小鼠(Nestin-Cre; Mark2+/+)存在显著差异:活动起始时间提前(ZT 12.05 ± 0.09 vs 12.37 ± 0.09,图7A,C),周期长度缩短约0.2小时(23.76 ±0.02 vs 23.96 ± 0.03,图7A,D)。虽然Nestin-Cre; Mark2flox/+和Nestin-Cre; Mark2flox/flox小鼠的运动活动均有类似程度减少(图7B),但活动起始时间提前和周期缩短仅见于Nestin-Cre; Mark2flox/ flox小鼠,表明该生物钟表型独立于运动活动变化(图7C)。因此,神经元特异性Mark2敲除小鼠的活动起始时间提前和周期缩短表型得到证明。
正常及神经元特异性Mark3基因敲除小鼠(图S11B)尽管运动活动减少(图7F,J),但活动起始时间(图7G,K)和生物钟周期(图7H,L)均无显著改变。此结果与D15细胞中的发现一致,表明Mark3不参与生物钟调节。

图7.神经元中Mark2(而非Mark3)生理性调控生物钟
(A-D)NestinCre/+;Mark2+/+(n=6)、NestinCre/+;Mark2+/flox(n=6)及NestinCre/+;Mark2flox/flox(n=7)小鼠节律的跑轮活动分析。灰色阴影区域表示黑暗(关灯)。代表性跑轮活动图(A),光照-黑暗循环条件下每10分钟跑轮转数(B),光照-黑暗循环条件下的活动起始时间(C)及持续黑暗条件下的生物钟周期长度(D)。(E-H)Mark3+/+(n=8)、Mark3+/-(n=8)及Mark3-/-(n=9)小鼠的代表性跑轮活动图。Mark3敲除小鼠虽活动量减少(F),但其活动起始时间(G)及周期长度(H)均无显著变化。(I-L)NestinCre/+;Mark3+/+(n=7)、NestinCre/+;Mark3+/flox(n=6)及NestinCre/+;Mark3flox/flox(n=6)小鼠节律的跑轮活动分析。神经元特异性Mark3敲除小鼠未见显著节律表型。统计分析采用单因素方差分析-Tukey多重比较检验(C,D,G,H,K,L);数据以均值±标准误表示。相关数据参见附图S11及附表S4。
鉴于Mark4敲除在D15细胞中表现出轻度周期缩短(图S10 J,K),我们同样构建了Mark4条件性敲除小鼠(图S11C),发现无节律变化(图S11D-G)。
综上所述,基因敲除小鼠研究结果显示,仅Mark2(而非Mark3或Mark4)突变体表现出与人类FASPS相似的活动起始时间提前和生物钟周期缩短表型⁴³‚⁶¹,证明Mark2对生物钟的调节至关重要。
讨论
在发现人类PER2 S662G突变与FASP综合征相关的25年后⁴⁸,本研究鉴定出MARK2作为PER2 S662的上游激酶,不仅能在体外生化水平磷酸化PER2 S662,更能在体内以S662依赖性方式参与细胞节律调节。在寻找PER2 S662上游激酶的过程中,我们发现7种可在生化水平磷酸化PER2 S662的激酶,但其各自敲除小鼠未表现出与人类S662G突变一致的活动起始时间提前表型。虽然不能排除内源性存在多种激酶共同调节PER2 S662的可能性,但本研究充分展示了生化纯化方法在解决生理学难题中的独特价值。
体外多种激酶对PER2 S662的磷酸化
自FASPS中发现hPER2 S662G突变以来⁴³‚⁶¹,寻找磷酸化S662的激酶一直是研究热点。文献报道⁴¹‚⁴³‚⁴⁶‚⁵¹‚⁵⁷⁻⁵⁹‚⁸⁹‚⁹³及本团队早期工作均发现CK1δ/ε是磷酸化S662的关键激酶。然而,在HEK293细胞中同时敲除CK1δ/ε虽显著降低但未消除S662磷酸化,这促使我们采用生化纯化方法,从而发现MARK2/3是S662激酶,二者属AMPK相关激酶家族⁶⁵⁻⁷⁷。通过对所有ARK家族成员对PER2 S662的体外活性进行比较(图4J),我们发现多个ARK成员可在体外磷酸化PER2 S662,包括SIK1-3、MARK1-4、TSSK1/2,而TSSK6、SNRK、MELK、AMPK1/2、NUAK1/2、BRSK1/2及HUNK则无此活性(图4)。然而,小鼠中敲低SIK3延长周期⁷⁸,而本团队既往发表的SIK1/2敲除⁷⁹‚⁸⁰及本文展示的TSSK1/2单敲/双敲(图S4)均未显示显著生物钟变化。这些看似矛盾的发现提示,存在多种S662激酶,可能在S662磷酸化中功能冗余,或在体内对其他生物钟组分发挥相反作用。
体内MARK2的功能意义
在四个MARK家族成员中,D15细胞过表达任一成员均可致周期延长(图6,S10);敲除MARK2/4缩短周期(图6,S10),而敲除MARK1/3在D15细胞中均无细胞节律表型。
MARK2敲除的生化和细胞节律表型在PER2 S662G突变D15细胞中消除(图6 G-H),此结果不仅支持MARK2在细胞节律调节中的作用,更表明MARK2作用于PER2 S662的直接上游。
证明MARK2生理重要性的关键证据来自敲除小鼠的研究。神经元特异性Mark2敲除小鼠同时表现出活动起始时间提前和周期长度缩短(约0.2小时,图7 A-D),而常规及神经元特异性Mark3敲除小鼠、神经元特异性Mark4敲除小鼠均无生物钟表型。因此,本研究最终发现Mark2是以S662依赖性方式调节生物钟的生理相关性激酶。
不能排除多种激酶(包括本研究发现)与MARK2在调节PER2 S662磷酸化和生物钟方面存在功能冗余的可能性。这些激酶可能调节PER2的多个位点,以及参与生物钟调节的其他蛋白的更多位点。
MARK2、PER2磷酸化与稳定性
翻译后修饰(特别是磷酸化)在生物钟调节中具有重要作用,在果蝇和哺乳动物中,PER蛋白发生磷酸化,磷酸化缺失会导致生物钟紊乱³⁶‚⁴⁰‚⁴¹‚⁴³‚⁴⁶‚⁴⁸‚⁵⁰‚⁵¹‚⁵⁶⁻⁶⁰‚⁶⟡‚⁶³‚⁸⁹‚⁹⁴⁻¹⟡⟡。
不同位点及不同蛋白的磷酸化对生物钟具有不同调节作用。就S662位点而言,其磷酸化可稳定PER2蛋白⁴¹‚⁴³‚⁴⁶,本研究证实了此点(图5 E,F)。此外,MARK2增强PER2稳定性,且此效应依赖于S662,因为MARK2无法稳定S662G突变型PER2(图5 G,H)。
近期研究显示,高脂饮食可增强PER2 S662位点磷酸化,而禁食则降低其磷酸化并导致行为节律时相提前¹⟡³。鉴于MARKs及其他ARKs可被上游激酶LKB1激活¹⟡⁴⁻¹⟡⁷,LKB1活性受多种刺激调节(高渗透压和APP积累激活,短期禁食则抑制)¹⟡⁸‚¹⟡⁹,探究LKB1-ARK信号轴是否作为整合器,通过调节PER2 S662磷酸化将代谢信息转化为生物钟计时信号,将是一个极具价值的研究方向。
通过生化方法揭示生物钟调节分子机制
遗传学方法在生物钟分子研究中具有重要价值。自果蝇¹³、脉孢菌¹⁴和衣藻¹⁵中发现遗传突变体以来,该方法已成为研究范式。但遗传学方法需要以整个动物为对象来发现参与生物钟调节的基因,这在低等生物中相对容易,在哺乳动物中则难度较大。
生化方法在鉴定已知参与生物钟调节蛋白方面更具优势,但体外生化活性与体内功能重要性之间并非必然关联。生化方法通常(虽非绝对)比遗传学方法更接近机制性理解。本研究中,MARK2通过磷酸化PER2 S662并稳定PER2蛋白,确立了其在PER2上游的作用位置。
综上所述,本研究采用蛋白质生化纯化方法,成功发现了可磷酸化PER2 S662的激酶,并从中证实小鼠脑内Mark2在生物钟调节中具有重要生理功能。
研究的局限性
本研究生化纯化方法虽有效,但所用HEK293细胞裂解液可能未能完全代表主时钟SCN的内源性激酶谱。Nestin-Cre驱动的敲除策略虽可移除神经元中的Mark2,但视交叉上核由异质性细胞群体(如表达AVP、VIP、GRP或CCK的神经元及胶质细胞)组成,需在这些亚群中进行细胞类型特异性敲除以更精准定义MARK2功能。此外,尽管CK1δ/ε是公认的PER2 S662激酶,MARK2活性如何受上游信号调节,及其是否与CK1δ/ε协同调节生物钟,目前尚不清楚。本研究还鉴定了其他数种可在体外磷酸化PER2 S662的激酶(SIK1-3,TSSK1/2/4,NIM1K),其在体内的生理作用及功能情境仍有待深入研究。
资源可用性
主要联系人
有关资源和试剂的进一步信息及请求请联系主要联系人饶毅(yrao@pku.edu.cn)。
材料可用性
本研究所产生的所有独特/稳定试剂均可无限制地从主要联系人处获取。
数据和代码可用性
本文报告的数据可根据要求由主要联系人共享。
本文未报告原始代码。
重新分析本文数据所需的任何附加信息均可根据要求从主要联系人处获取。
致谢
感谢北京大学国家蛋白质科学中心的刘栋博士和张琪博士在质谱样品准备和数据分析中的帮助,感谢北京大学国家蛋白质科学中心提供的仪器使用便利。
作者贡献
饶毅指导;饶毅、刘玉祥、李扬、王涛、余腾辉共同设计实验;饶毅、刘玉祥、李扬、王涛论文初稿,并整合所有合作者数据;刘玉祥完成生化纯化和体外测定实验;李扬完成小鼠节律实验;王涛完成D15细胞实验;余腾辉完成phos-tag cDNA筛选实验;黄娟负责基因突变小鼠的构建。
利益冲突声明
作者声明无竞争性利益。
STAR★方法
本文的详细方法可在在线版本中获取,并包含以下内容:
关键资源表
实验模型和研究对象详细信息
细胞系
动物
方法细节
Phos-tag检测磷酸化
重组蛋白的表达与纯化
从HEK293T细胞中纯化MARK2和MARK3
体外激酶测定
米氏常数计算
蛋白质稳定性测定
敲除细胞系的构建
D15细胞的节律分析
使用Prime Editing 7生成PER2 S662G突变D15细胞
小鼠品系
小鼠节律的跑轮测定
定量与统计分析
补充信息
补充信息可在在线获取:https://doi.org/10.1016/j.chembiol.2026.02.007
投稿日期:2025年7月24日
修订日期: 2025年11月18日
接受日期:2026年2月17日
参考文献
1.Roenneberg, T., and Merrow, M. (2016). The Circadian Clock and Human Health. Current Biology 26, R432-443.
2.Parsons, M.J., Moffitt, T.E., Gregory, A.M., Goldman-Mellor, S., Nolan, P.M., Poulton, R., and Caspi, A. (2015). Social jetlag, obesity and metabolic disorder: investigation in a cohort study. Int J Obes (Lond) 39, 842-848.
3.Ohlander, J., Keskin, M.C., Stork, J., and Radon, K. (2015). Shift work and hypertension: Prevalence and analysis of disease pathways in a German car manufacturing company. American Journal of Industrial Medicine 58, 549-560.
4.Roenneberg, T., Allebrandt, K.V., Merrow, M., and Vetter, C. (2012). Social jetlag and obesity. Current Biology 22, 939-943.
5.Schernhammer, E.S., Laden, F., Speizer, F.E., Willett, W.C., Hunter, D.J., Kawachi, I., Fuchs, C.S., and Colditz, G.A. (2003). Night-shift work and risk of colorectal cancer in the nurses' health study. J Natl Cancer Inst 95, 825-828.
6.Tynes, T., Hannevik, M., Andersen, A., Vistnes, A.I., and Haldorsen, T. (1996). Incidence of breast cancer in Norwegian female radio and telegraph operators. Cancer Causes & Control 7, 197-204.
7.Azmi, N.A.S.M., Juliana, N., Teng, N.I.M.F., Azmani, S., Das, S., and Effendy, N. (2020). Consequences of Circadian Disruption in Shift Workers on Chrononutrition and their Psychosocial Well-Being. Int J Env Res Pub He 17.
8.Curtis, A.M., Bellet, M.M., Sassone-Corsi, P., and O'Neill, L.A.J. (2014). Circadian Clock Proteins and Immunity. Immunity 40, 178-186.
9.Chong, S.Y., Ptacek, L.J., and Fu, Y.H. (2012). Genetic insights on sleep schedules: this time, it's PERsonal. Trends in genetics : TIG 28, 598-605.
10.Bass, J., and Takahashi, J.S. (2010). Circadian Integration of Metabolism and Energetics. Science 330, 1349-1354.
11.Mohawk, J.A., and Takahashi, J.S. (2011). Cell autonomy and synchrony of suprachiasmatic nucleus circadian oscillators. Trends Neurosci 34, 349-358.
12.Dibner, C., Schibler, U., and Albrecht, U. (2010). The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annual Review of Physiology 72, 517-549.
13.Konopka, R.J., and Benzer, S. (1971). Clock mutants of Drosophila melanogaster. Proceedings of the National Academy of Science USA. 68, 2112-2116.
14.Feldman, J.F., and Wasar, N. (1971). in Biochronometry. National Academy of Science, Washington DC, 652-656.
15.Bruce, V.G. (1972). Mutants of the biological clock in Chlamydomonas reinhardi. J Genetics. 70, 537-548.
16.Lowrey, P.L., and Takahashi, J.S. (2011). Genetics of Circadian Rhythms in Mammalian Model Organisms. Adv Genet 74, 175-230.
17.Crane, B.R., and Young, M.W. (2014). Interactive features of proteins composing eukaryotic circadian clocks. Annual Review of Biochemistry 83, 191-219.
18.Panda, S., Hogenesch, J.B., and Kay, S.A. (2002). Circadian rhythms from flies to human. Nature 417, 329-335.
19.Reppert, S.M., and Weaver, D.R. (2001). Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol 63, 647-676.
20.Allada, R., Emery, P., Takahashi, J.S., and Rosbash, M. (2001). Stopping time: the genetics of fly and mouse circadian clocks. Annu Rev Neurosci 24, 1091-1119.
21.Hardin, P.E., Hall, J.C., and Rosbash, M. (1990). Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature 343, 536-540.
22.Zheng, X., and Sehgal, A. (2012). Speed control: cogs and gears that drive the circadian clock. Trends in Neurosciences 35, 574-585.
23.Nitabach, M.N., and Taghert, P.H. (2008). Organization of the Drosophila circadian control circuit. Current Biology 18, R84-93.
24.Vitaterna, M.H., Shimomura, K., and Jiang, P. (2019). Genetics of Circadian Rhythms. Neurol Clin 37, 487-504.
25.Shearman, L.P., Sriram, S., Weaver, D.R., Maywood, E.S., Chaves, I., Zheng, B.H., Kume, K., Lee, C.C., van der Horst, G.T.J., Hastings, M.H. et al. (2000). Interacting molecular loops in the mammalian circadian clock. Science 288, 1013-1019.
26.Zheng, B., Larkin, D.W., Albrecht, U., Sun, Z.S., Sage, M., Eichele, G., Lee, C.C., and Bradley, A. (1999). The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature 400, 169-173.
27.Kume, K., Zylka, M.J., Sriram, S., Shearman, L.P., Weaver, D.R., Jin, X.W., Maywood, E.S., Hastings, M.H., and Reppert, S.M. (1999). mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98, 193-205.
28.Hogenesch, J.B., Gu, Y.Z., Jain, S.J., and Bradfield, C.A. (1998). The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci USA 95, 5474-5479.
29.Gekakis, N., Staknis, D., Nguyen, H.B., Davis, F.C., Wilsbacher, L.D., King, D.P., Takahashi, J.S., and Weitz, C.J. (1998). Role of the CLOCK protein in the mammalian circadian mechanism. Science 280, 1564-1569.
30.Shearman, L.P., Zylka, M.J., Weaver, D.R., Kolakowski, L.F., Jr., and Reppert, S.M. (1997). Two period homologs: circadian expression and photic regulation in the suprachiasmatic nuclei. Neuron 19, 1261-1269.
31.Chen, R.M., Schirmer, A., Lee, Y., Lee, H., Kumar, V., Yoo, S.H., Takahashi, J.S., and Lee, C. (2009). Rhythmic PER Abundance Defines a Critical Nodal Point for Negative Feedback within the Circadian Clock Mechanism. Mol Cell 36, 417-430.
32.Lee, H., Chen, R.M., Lee, Y., Yoo, S., and Lee, C. (2009). Essential roles of CKIδ and CKIε in the mammalian circadian clock. Proc Natl Acad Sci USA 106, 21359-21364.
33.Cao, X.M., Yang, Y.Y., Selby, C.P., Liu, Z.X., and Sancar, A. (2021). Molecular mechanism of the repressive phase of the mammalian circadian clock. Proc Natl Acad Sci USA 118.
34.Aryal, R.P., Kwak, P.B., Tamayo, A.G., Gebert, M., Chiu, P.L., Walz, T., and Weitz, C.J. (2017). Macromolecular Assemblies of the Mammalian Circadian Clock. Mol Cell 67, 770.
35.Park, J., Lee, K.J., Kim, H., Shin, H., and Lee, C.G. (2023). Endogenous circadian reporters reveal functional differences of paralogs and the significance of PERIOD:CK 1 stable interaction. Proc Natl Acad Sci USA 120.
36.An, Y., Yuan, B.S., Xie, P.C., Gu, Y., Liu, Z.W., Wang, T., Li, Z.H., Xu, Y., and Liu, Y. (2022). Decoupling PER phosphorylation, stability and rhythmic expression from circadian clock function by abolishing PER-CK1 interaction. Nat Commun 13.
37.Preitner, N., Damiola, F., Lopez-Molina, L., Zakany, J., Duboule, D., Albrecht, U., and Schibler, U. (2002). The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110, 251-260.
38.Liao, M., Liu, Y., Xu, Z., Fang, M., Yu, Z., Cui, Y., Sun, Z., Huo, R., Yang, J., Huang, F. et al. (2025). The P-loop NTPase RUVBL2 is a conserved clock component across eukaryotes. Nature. 10.1038.
39.Lee, C., Etchegaray, J.P., Cagampang, F.R., Loudon, A.S., and Reppert, S.M. (2001). Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 107, 855-867.
40.Chen, R. (2009). Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Mol. Cell. 36, 417-430.
41.Vanselow, K., Vanselow, J.T., Westermark, P.O., Reischl, S., Maier, B., Korte, T., Herrmann, A., Herzel, H., Schlosser, A., and Kramer, A. (2006). Differential effects of PER2 phosphorylation: molecular basis for the human familial advanced sleep phase syndrome (FASPS). Gene Dev 20, 2660-2672.
42.Fu, Y., Jones, C.R., Toh, K., Virshup, D., and Ptacek, L.J. (2001). An hPer2 phosphorylation site mutation in familial Advanced Sleep-Phase Syndrome. Am J Hum Genet 69, 597-597.
43.Xu, Y., Toh, K.L., Jones, C.R., Shin, J.Y., Fu, Y.H., and Ptácek, L.J. (2007). Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell 128, 59-70.
44.Ohsaki, K., Oishi, K., Kozono, Y., Nakayama, K., Nakayama, K.I., and Ishida, N. (2008). The Role of β-TrCP1 and β-TrCP2 in Circadian Rhythm Generation by Mediating Degradation of Clock Protein PER2. J Biochem 144, 609-618.
45.Cao, X.M., Wang, L., Selby, C.P., Lindsey-Boltz, L.A., and Sancar, A. (2023). Analysis of mammalian circadian clock protein complexes over a circadian cycle. J Biol Chem 299.
46.Xu, Y., Padiath, Q.S., Shapiro, R.E., Jones, C.R., Wu, S.C., Saigoh, N., Saigoh, K., Ptácek, L.J., and Fu, Y.H. (2005). Functional consequences of a CKI δ mutation causing familial advanced sleep phase syndrome. Nature 434, 640-644.
47.Flotow, H., Graves, P.R., Wang, A.Q., Fiol, C.J., Roeske, R.W., and Roach, P.J. (1990). Phosphate groups as substrate determinants for casein kinase I action. J. Biol. Chem. 265, 14264-14269.
48.Toh, K.L. (2001). An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. J Science. 291, 1040-1043.
49.Gallego, M., and Virshup, D.M. (2007). Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol 8, 139-148.
50.Zhou, M., Kim, J.K., Eng, G.W., Forger, D.B., and Virshup, D.M. (2015). A Period2 phosphoswitch regulates and temperature compensates circadian period. Mol Cell. 60, 77-88.
51.Narasimamurthy, R., Hunt, S.R., Lu, Y.N., Fustin, J.M., Okamura, H., Partch, C.L., Forger, D.B., Kim, J.K., and Virshup, D.M. (2018). CK1δ/ε protein kinase primes the PER2 circadian phosphoswitch. P Natl Acad Sci USA 115, 5986-5991.
52.Masuda, S. (2020). Mutation of a PER2 phosphodegron perturbs the circadian phosphoswitch. Proc. Natl Acad. Sci. USA. 117, 10888-10896.
53.Cullati, S., Akizuki, K., Chen, J.S., Johnson, J., Yaron, T., Cantley, L., and Gould, K. (2024). Substrate displacement of CK1 C-termini regulates kinase specificity. J Biol Chem 300, S706-S706.
54.Francisco, J.C., and Virshup, D.M. (2024). Hierarchical and scaffolded phosphorylation of two degrons controls PER2 stability. J Biol Chem 300.
55.Francisco, J.C., and Virshup, D.M. (2022). Casein Kinase 1 and Human Disease: Insights From the Circadian Phosphoswitch. Front Mol Biosci 9.
56.Ricci, C.G., Philpott, J.M., Torgrimson, M.R., Freeberg, A.M., Narasimamurthy, R., Pécora de Barros, E., Amaro, R., Virshup, D.M., McCammon, J.A., and Partch, C.L. (2025). Markovian State Models uncover Casein Kinase 1 dynamics that govern circadian period. 2025.2001.2017.633651.
57.Philpott, J.M., Freeberg, A.M., Park, J., Lee, K.J., Ricci, C.G., Hunt, S.R., Narasimamurthy, R., Segal, D.H., Robles, R., Cai, Y. et al. (2023). PERIOD phosphorylation leads to feedback inhibition of CK1 activity to control circadian period. Mol Cell 83, 1677-1692.
58.Philpott, J.M., Narasimamurthy, R., Ricci, C.G., Freeberg, A.M., Hunt, S.R., Yee, L.E., Pelofsky, R.S., Tripathi, S., Virshup, D.M., and Partch, C.L. (2020). Casein kinase 1 dynamics underlie substrate selectivity and the PER2 circadian phosphoswitch. Elife 9.
59.Philpott, J.M., Torgrimson, M.R., Harold, R.L., and Partch, C.L. (2022). Biochemical mechanisms of period control within the mammalian circadian clock. Semin Cell Dev Biol 126, 71-78.
60.Shanware, N.P., Hutchinson, J.A., Kim, S.H., Zhan, L., Bowler, M.J., and Tibbetts, R.S. (2011). Casein kinase 1-dependent phosphorylation of familial advanced sleep phase syndrome-associated residues controls PERIOD 2 stability. J Biol Chem. 286, 12766-12774.
61.Toh, K.L., Jones, C.R., He, Y., Eide, E.J., Hinz, W.A., Virshup, D.M., Ptácek, L.J., and Fu, Y.H. (2001). An hPer2 phosphorylation site mutation in familiar advanced sleep phase syndrome. Science 291, 1040-1043.
62.Eide, E.J. (2005). Control of mammalian circadian rhythm by CKI epsilon regulated proteasome-mediated PER2 degradation. Mol. Cell. Biol. 25, 2795-2807.
63.Masuda, S., Narasimamurthy, R., Yoshitane, H., Kim, J.K., Fukada, Y., and Virshup, D.M. (2020). Mutation of a PER2 phosphodegron perturbs the circadian phosphoswitch. Proc Natl Acad Sci USA 117, 10888-10896.
64.Fustin, J.M., Kojima, R., Itoh, K., Chang, H.Y., Ye, S.Q., Zhuang, B.W., Oji, A., Gibo, S., Narasimamurthy, R., Virshup, D. et al. (2018). Two δ transcripts regulated by m6A methylation code for two antagonistic kinases in the control of the circadian clock. Proc Natl Acad Sci USA 115, 5980-5985.
65.Beg, Z.H., Allmann, D.W., and Gibson, D.M. (1973). Modulation of 3-hydroxy-3-methylglutaryl coenzyme: a reductase activity with cAMP and with protein fractions of rat liver cytosol.Biochem Biophys Res Comm. 54, 1362-1369.
66.Carlson, C.A., and Kim, K.H. (1973). Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J Biol Chem. 248, 378-380.
67.Ingebritsen, T.S., Lee, H., Parker, R.A., and Gibson, D.M. (1978). Reversible modulation of the activities of both liver microsomal hydroxymethylglutaryl coenzyme A reductase and its inactivating enzyme. Evidence for regulation by phosphorylation-dephosphorylation. Biochem Biophys Res Comm. 81, 1268-1277.
68.Yeh, L.A., Lee, K.H., and Kim, K.H. (1980). Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by the adenylate energy charge. J Biol Chem. 255, 2308-2314.
69.Ferrer, A., Caelles, C., Massot, N., and Hegardt, F.G. (1985). Activation of rat liver cytosolic 3-hydroxy-3-methylglutaryl coenzyme A reductase kinase by adenosine 5'-monophosphate. Biochem Biophys Res Comm. 132, 497-504.
70.Carling, D., Zammit, V.A., and Hardie, D.G. (1987). A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 223, 217-222.
71.Munday, M.R., Campbell, D.G., Carling, D., and Hardie, D.G. (1988). Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem. 175, 331-338.
72.Carling, D., Clarke, P.R., Zammit, V.A., and Hardie, D.G. (1989). Purification and characterization of the AMP-activated protein kinase. Co-purification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-Co-A reductase kinase activities. Eur J Biochem. 186, 129-136.
73.Hardie, D.G. (2014). AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annu Rev Nutr. 34, 31-55.
74.López, M., Nogueiras, R., Tena-Sempere, M., and Diéguez, C. (2016). Hypothalamic AMPK: a canonical regulator of whole-body energy balance. Nat Rev Endocrinol. 12, 421-432.
75.Hardie, D.G., Schaffer, B.E., and Brunet, A. (2016). AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26, 190-201.
76.Herzig, S., and Shaw, R.J. (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 19, 122-135.
77.Liu, Y.X., Wang, T.V., Cui, Y.F., Li, C.Y., Jiang, L.F., and Rao, Y. (2022). STE20 phosphorylation of AMPK related kinases revealed by biochemical purifications combined with genetics. J Biol Chem. 298, 101928.
78.Hayasaka, N., Hirano, A., Miyoshi, Y., Tokuda, I.T., Yoshitane, H., Matsuda, J., and Fukada, Y. (2017). Salt-inducible kinase 3 regulates the mammalian circadian clock by destabilizing PER2 protein. Elife. 6, e24779.
79.Yu, J.J., Liu, H.J., Gao, R., Wang, T.V., Li, C.G., Liu, Y.X., Yang, L., Xu, Y., Cui, Y.F., Jia, C.X. et al. (2025). Calcineurin: An essential regulator of sleep revealed by biochemical, chemical biological, and genetic approaches. Cell Chem Biol 32, 157-173.
80.Li, Y., Li, C.A., Liu, Y.X., Yu, J.J., Yang, J.Q., Cui, Y.F., Wang, T., Li, C.Y., Jiang, L.F., Song, M.L. et al. (2023). Sleep need, the key regulator of sleep homeostasis, is indicated and controlled by phosphorylation of threonine 221 in salt-inducible kinase 3. Genetics 225.
81.Akashi, M., Tsuchiya, Y., Yoshino, T., and Nishida, E. (2002). Control of intracellular dynamics of mammalian period proteins by casein kinase I ε and CKIδ in cultured cells. Mol Cell Biol. 22, 1693-1703.
82.Kinoshita, E., Kinoshita-Kikuta, E., Takiyama, K., and Koike, T. (2006). Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteom. 5, 749-757.
83.Zhang, E.E., Liu, A.C., Hirota, T., Miraglia, L.J., Welch, G., Pongsawakul, P.Y., Liu, X., Atwood, A., Huss, J.W., 3rd, Janes, J. et al. (2009). A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell 139, 199-210.
84.Eide, E.J., Woolf, M.F., Kang, H., Woolf, P., Hurst, W., Camacho, F., Vielhaber, E.L., Giovanni, A., and Virshup, D.M. (2005). Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol 25, 2795-2807.
85.Lowrey, P.L., Shimomura, K., Antoch, M.P., Yamazaki, S., Zemenides, P.D., Ralph, M.R., Menaker, M., and Takahashi, J.S. (2000). Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science 288, 483-492.
86.Maier, B., Wendt, S., Vanselow, J.T., Wallach, T., Reischl, S., Oehmke, S., Schlosser, A., and Kramer, A. (2009). A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes Dev 23, 708-718.
87.Iitaka, C., Miyazaki, K., Akaike, T., and Ishida, N. (2005). A role for glycogen synthase kinase-3 beta in the mammalian circadian clock. J Biol Chem 280, 29397-29402.
88.Honda, T., Fujiyama, T., Miyoshi, C., Ikkyu, A., Hotta-Hirashima, N., Kanno, S., Mizuno, S., Sugiyama, F., Takahashi, S., and Funato, H. (2018). A single phosphorylation site of SIK3 regulates daily sleep amounts and sleep need in mice. Proc Natl Acad Sci USA. 115, 10458-10463.
89.Meng, Q.J., Logunova, L., Maywood, E.S., Gallego, M., Lebiecki, J., Brown, T.M., Sládek, M., Semikhodskii, A.S., Glossop, N.R.J., Piggins, H.D. et al. (2008). Setting clock speed in mammals:: The CK1ε tau mutation in mice accelerates circadian pacemakers by selectively destabilizing PERIOD proteins. Neuron 58, 78-88.
90.Kolodziej, P.A., and Young, R.A. (1991). Epitope tagging and protein surveillance. Methods Enzymol. 194, 508-519.
91.Liu, Y.X., Wang, T.V., Cui, Y.F., Gao, S.X., and Rao, Y. (2022). Biochemical purification uncovers mammalian sterile 3 (MST3) as a new protein kinase for multifunctional protein kinases AMPK and SIK3. J Biol Chem. 298, 101929.
92.Jagannath, A. (2013). The CRTC1-SIK1 pathway regulates entrainment of the circadian clock. Cell 154, 1100-1111.
93.Hirota, T., Lee, J.W., Lewis, W.G., Zhang, E.E., Breton, G., and Liu, X. (2010). High-throughput chemical screen identifies a novel potent modulator of cellular circadian rhythms and reveals CKIalpha as a clock regulatory kinase. PLoS Biol. 8, e1000559.
94.Edery, I., Zwiebel, L.J., Dembinska, M.E., and Rosbash, M. (1994). Temporal phosphorylation of the Drosophila period protein. Proc Natl Acad Sci USA. 91, 2260-2264.
95.Kloss, B., Price, J.L., Saez, L., Blau, J., Rothenfluh, A., and Wesley, C.S. (1998). The Drosophila clock gene double-time encodes a protein closely related to human casein kinase Iepsilon. Cell 94, 97-107.
96.Price, J.L., Blau, J., Rothenfluh, A., Abodeely, M., Kloss, B., and Young, M.W. (1998). double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. J Cell. 94, 83-95.
97.Vielhaber, E., Eide, E., Rivers, A., Gao, Z.H., and Virshup, D.M. (2000). Nuclear entry of the circadian regulator mPER1 is controlled by mammalian casein kinase I ε. Mol Cell Biol 20, 4888-4899.
98.Kloss, B., Rothenfluh, A., Young, M.W., and Saez, L. (2001). Phosphorylation of period is influenced by cycling physical associations of double-time, period, and timeless in the Drosophila clock. Neuron 30, 699-706.
99.Lee, C., Weaver, D.R., and Reppert, S.M. (2004). Direct association between mouse PERIOD and CKIε is critical for a functioning circadian clock. Mol Cell Biol. 24, 584-594.
100.Miyazaki, K., Nagase, T., Mesaki, M., Narukawa, J., Ohara, O., and Ishida, N. (2004). Phosphorylation of clock protein PER1 regulates its circadian degradation in normal human fibroblasts. Biochem J. 380, 95-103.
101.Nawathean, P., and Rosbash, M. (2004). The doubletime and CKII kinases collaborate to potentiate Drosophila PER transcriptional repressor activity. Mol Cell. 10, 213-223.
102.Sathyanarayanan, S., Zheng, X., Xiao, R., and Sehgal, A. (2004). Posttranslational regulation of Drosophila PERIOD protein by protein phosphatase 2A. Cell 116, 603-615.
103.Takano, A., Isojima, Y., and Nagai, K. (2004). Identification of mPer1 phosphorylation sites responsible for the nuclear entry. J Biol Chem 279, 32578-32585.
104.Cyran, S.A., Yiannoulos, G., Buchsbaum, A.M., Saez, L., Young, M.W., and Blau, J. (2005). The double-time protein kinase regulates the subcellular localization of the Drosophila clock protein period. J Neurosci. 25, 5430-5437.
105.Lin, J.M., Schroeder, A., and Allada, R. (2005). In vivo circadian function of casein kinase 2 phosphorylation sites in Drosophila PERIOD. J Neurosci. 25, 11175-11183.
106.Shirogane, T., Jin, J., Ang, X.L., and Harper, J.W. (2005). SCFbeta-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J Biol Chem. 280, 26863-26872.
107.Gallego, M., Kang, H., and Virshup, D.M. (2006). Protein phosphatase 1 regulates the stability of the circadian protein PER2. Biochem J. 399, 169-175.
108.He, Q. (2006). CKI and CKII mediate the FREQUENCY-dependent phosphorylation of the WHITE COLLAR complex to close the Neurospora circadian negative feedback loop. Genes Dev. 20, 2552-2565.
109.Kim, E.Y., Ko, H.W., Yu, W., Hardin, P.E., and Edery, I. (2007). A DOUBLETIME kinase binding domain on the Drosophila PERIOD protein is essential for its hyperphosphorylation, transcriptional repression, and circadian clock function. Mol Cell Biol. 27, 5014-5028.
110.Nawathean, P., Stoleru, D., and Rosbash, M. (2007). A small conserved domain of Drosophila PERIOD is important for circadian phosphorylation, nuclear localization, and transcriptional repressor activity. Molecular Cell Biol. 27, 5002-5013.
111.Blau, J. (2008). PERspective on PER phosphorylation. J Genes Dev. 22, 1737-1740.
112.Chiu, J.C., Vanselow, J.T., Kramer, A., and Edery, I. (2008). The phospho-occupancy of an atypical SLIMB-binding site on PERIOD that is phosphorylated by DOUBLETIME controls the pace of the clock. Genes Dev. 22, 1758-1772.
113.Etchegaray, J.P., Machida, K.K., Noton, E., Constance, C.M., Dallmann, R., and Di Napoli, M.N. (2009). Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol Cell Biol. 29, 3853-3866.
114.Ko, H.W., Kim, E.Y., Chiu, J., Vanselow, J.T., Kramer, A., and Edery, I. (2010). A hierarchical phosphorylation cascade that regulates the timing of PERIOD nuclear entry reveals novel roles for proline-directed kinases and GSK-3beta/SGG in circadian clocks. J Neurosci. 30, 12664-12675.
115.Schmutz, I., Wendt, S., Schnell, A., Kramer, A., Mansuy, I.M., and Albrecht, U. (2011). Protein phosphatase 1 (PP1) is a post-translational regulator of the mammalian circadian clock. PloS One. 6, e21325.
116.Chiu, J.C., Ko, H.W., and Edery, I. (2011). NEMO/NLK phosphorylates PERIOD to initiate a time-delay phosphorylation circuit that sets circadian clock speed. Cell 145, 357-370.
117.Lee, H.M., Chen, R.M., Kim, H., Etchegaray, J.P., Weaver, D.R., and Lee, C. (2011). The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc Natl Acad Sci USA 108, 16451-16456.
118.Uchida, Y., Osaki, T., Yamasaki, T., Shimomura, T., Hata, S., and Horikawa, K. (2012). Involvement of stress kinase mitogen-activated protein kinase kinase 7 in regulation of mammalian circadian clock. J Biol Chem. 287, 8318-8326.
119.Garbe, D.S., Fang, Y., Zheng, X., Sowcik, M., Anjum, R., and Gygi, S.P. (2013). Cooperative interaction between phosphorylation sites on PERIOD maintains circadian period in Drosophila. Plos Genet. 9, e1003749.
120.Kaasik, K., Kivimae, S., Allen, J.J., Chalkley, R.J., Huang, Y., and Baer, K. (2013). Glucose Sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian Clock. Cell Metab. 17, 291-302.
121.Maywood, E.S., Chesham, J.E., Smyllie, N.J., and Hastings, M.H. (2014). The Tau mutation of casein kinase 1epsilon sets the period of the mammalian pacemaker via regulation of Period1 or Period2 clock proteins. J Biol Rhyth. 29, 110-118.
122.Marzoll, D. (2022). Casein kinase 1 and disordered clock proteins form functionally equivalent, phospho-based circadian modules in fungi and mammals. Proc. Natl Acad. Sci. USA. 119, e2118286119.
123.Levine, D.C., Reeh, R.H., McMahon, T., Mandrup-Poulsen, T., Fu, Y.-H., and Ptáček, L.J. (2025). Unsaturated fat alters clock phosphorylation to align rhythms to the season in mice. Science. eadp3065.
124.Lizcano, J.M., Göransson, O., Toth, R., Deak, M., Morrice, N.A., Boudeau, J., Hawley, S.A., Udd, L., Mäkelä, T.P., Hardie, D.G. et al. (2004). LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J 23, 833-843.
125.Jaleel, M., McBride, A., Lizcano, J.M., Deak, M., Toth, R., Morrice, N.A., and Alessi, D.R. (2005). Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 579, 1417-1423.
126.Thirugnanam, K., and Ramchandran, R. (2020). SNRK: a metabolic regulator with multifaceted role in development and disease. Vessel Plus. 4, 26.
127.Kim, C.L., Lim, S.B., and Choi, S.H. (2024). The LKB1--TSSK1B axis controls YAP phosphorylation to regulate the Hippo--YAP pathway. Cell Death Dis. 15, 76.
128.Wang, J.W., Imai, Y., and Lu, B.W. (2007). Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J Neurosci 27, 574-581.
129.Choi, S., Lim, D.S., and Chung, J. (2015). Feeding and Fasting Signals Converge on the LKB1-SIK3 Pathway to Regulate Lipid Metabolism in Drosophila. Plos Genet 11.
130.Sanjana, N.E., Shalem, O., and Zhang, F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783-784.
131.Zhang, E.E., Liu, A.C., Hirota, T., Miraglia, L.J., Welch, G., Pongsawakul, P.Y., Liu, X.Z., Atwood, A., Huss, J.W., Janes, J. et al. (2009). A Genome-wide RNAi Screen for Modifiers of the Circadian Clock in Human Cells. Cell 139, 199-210.
STAR★方法
关键资源表










实验模型和研究对象详细信息
细胞系
HEK293T(源自胎儿,雌性)细胞系购自ATCC(CRL-3216),信息可在ATCC官网查询。细胞培养于含10%胎牛血清及1%青霉素/链霉素的DMEM培养基中,置于37°C、5% CO₂培养箱内,每2天传代一次。D15(Per2::dLuc U2OS)细胞由北京生命科学研究所张二荃教授惠赠¹³¹。其亲本U2OS细胞系来源于一例15岁女性骨肉瘤患者,且已通过ATCC验证。该细胞维持于含10%胎牛血清、1%青霉素/链霉素及MEM非必需氨基酸的DMEM培养基中。本研究所用所有细胞系在实验前均经支原体检测且结果为阴性。
动物
所有动物实验程序均遵循相关指导原则,并获得北京脑科学与类脑研究中心动物护理与使用委员会的批准。突变小鼠及其野生型同窝对照均维持在C57BL/6N遗传背景上。小鼠饲养于温度及湿度可控的设施内,遵循12小时光照/12小时黑暗循环,可自由获取食物和水。
方法细节
Phos-tag检测磷酸化
Phos-tag SDS-PAGE参照制造商(Phos-tag Acrylamide AAL-107)方案进行,略作修改,凝胶中Phos-tag终浓度为25 μM。Phos-tag可与蛋白质上的磷酸基团结合,在SDS-PAGE过程中减缓其迁移速度⁸⟡。采用抗FLAG-辣根过氧化物酶抗体进行免疫印迹,检测磷酸化hPER2片段对应的迁移条带。
重组蛋白的表达与纯化
将目的基因克隆至带有指定标签的pET-28a载体中。将重组质粒转化至大肠杆菌BL21(DE3)感受态细胞。挑取单菌落接种于5 mL含50 μg/mL卡那霉素的LB培养基中,于37°C、220 rpm振荡培养过夜。将过夜培养物按1:1000稀释至含卡那霉素的新鲜LB培养基中,于37°C培养至OD600达0.6-0.8。加入0.5 mM异丙基-β-D-硫代半乳糖苷诱导蛋白表达,于18°C继续振荡培养16小时。收集菌体,重悬于含蛋白酶抑制剂的Ni2+结合缓冲液(300 mM NaCl,20 mM Tris-HCl,pH7.5)中,超声破碎后于14000 rpm离心30分钟。上清经Ni⟡⁺亲和层析柱纯化,纯化后的重组蛋白保存于-80°C。用作底物的蛋白保留其融合标签。用作激酶的蛋白,将融合蛋白与His标签的TEV蛋白酶共同孵育,以切MBP蛋白标签及GFP-8×His标签,释放未标记的激酶蛋白。酶切产物再次通过Ni-NTA柱,去除GFP-8×His标签、TEV蛋白酶及未酶切的融合蛋白。收集含未标记蛋白及MBP标签的流穿液,进一步通过MBP亲和层析去除残余的含MBP片段。最终纯化的未标记蛋白经离心超滤法置换至缓冲液A(20 mM HEPES pH 7.5,150 mM NaCl,1 mM DTT)中,液氮速冻后保存于-80°C。
从HEK293T细胞中纯化MARK2和MARK3
取500 ml HEK293T细胞裂解液(蛋白浓度10 mg/ml),按图1C所示流程依次通过连续连接的层析柱进行纯化。纯化过程在4°C条件下使用AKTA Purifier 10 FPLC系统(GE Healthcare)完成。将细胞裂解液上样至预先用缓冲液A平衡的10 ml SP HP柱,以20倍柱体积的0-600 mM NaCl线性梯度洗脱。收集20个组分,取各组分0.5 ml透析至缓冲液A后,进行体外PER2 S662磷酸化活性测定。合并SP HP柱活性组分(7和8)(图1D),上样至10 ml Blue HP柱,以20倍柱体积的0-3000 mM NaCl线性梯度洗脱。合并Blue柱活性组分(7-11,1050-1650 mM NaCl)(图1E),上样至10 ml Heparin HP柱,以20倍柱体积的0-600 mM NaCl线性梯度洗脱。合并Heparin HP柱活性组分(9-13,270-330 mM NaCl)(图1F),上样至10 ml Q HP柱,以20倍柱体积的0-600 mM NaCl线性梯度洗脱。合并Q HP柱活性组分(4-7,120-210 mM NaCl)(图1G),上样至1 ml HAP羟基磷灰石柱,以0-300 mM K₂PO₄线性梯度洗脱。合并HAP柱活性组分(11和12,150-180 mM K₂PO₄)(图1H),浓缩至0.5 ml后上样至Superdex 200 10/300凝胶过滤柱,以含200 mM NaCl的缓冲液A洗脱。测定Superdex 200柱各组分(图1I)的PER2 S662磷酸化活性,将活性组分(11和12,200 mM NaCl)进行质谱分析。
体外激酶活性测定
将纯化的分离组分(10 μl)或重组激酶(0.5 μg)与1 μg底物在缓冲液A(20 mM HEPES,10 mM KCl,1.5 mM MgCl₂,1 mM EDTA,1 mM EGTA,1 mM DTT,1×蛋白酶抑制剂混合物,1×磷酸酶抑制剂II,1×磷酸酶抑制剂III)中混合,加入1 mM ATP(pH7.5)至总体积20 μl,于37°C孵育1小时。加入蛋白上样缓冲液并于95°C加热终止反应,随后进行免疫印迹分析。
米氏常数测定
将0.2 μg重组激酶与不同浓度的底物在缓冲液A(20 mM HEPES,10 mM KCl,1.5 mM MgCl₂,1 mM EDTA,1 mM EGTA,1 mM DTT,1×蛋白酶抑制剂混合物,1×磷酸酶抑制剂II,1×磷酸酶抑制剂III)中混合,于37°C孵育20分钟。加入蛋白上样缓冲液并于95°C加热终止反应,进行免疫印迹分析。采用ImageJ软件对免疫印迹结果进行定量,并使用GraphPad Prism软件计算Km值。
蛋白质稳定性测定
将PER2野生型或S662G突变体质粒单独转染或与MARK2质粒共转染HEK293T细胞24小时。转染后细胞经200 μM放线菌酮处理,在指定时间点收集蛋白样品。等量蛋白提取物经指定抗体进行免疫印迹分析,以肌动蛋白为内参。采用ImageJ软件对免疫印迹结果进行定量,并使用GraphPad Prism软件计算蛋白质半衰期。
敲除细胞系的构建
采用CRISPR/Cas9技术构建MARK1-4基因敲除的HEK细胞。将靶向目的基因的两个串联sgRNA序列插入lentiCRISPRv2 puro载体。使用Lipofectamine 3000将lentiCRISPRv2质粒转染至细胞,经嘌呤霉素筛选后,采用有限稀释法在96孔板中挑选单克隆。通过相应抗体的免疫印迹验证单克隆细胞的基因敲除效果。
D15细胞节律分析
进行节律记录时,将细胞传代至10 mm × 35 mm培养皿中,待细胞融合度达100%时,更换为XM培养基(1×DMEM、1×B27添加剂、4.2 mM NaHCO₃、10 mM HEPES、100 U/ml青霉素-链霉素、1 mM荧光素)进行同步化处理。用高真空硅脂及显微镜盖玻片密封培养皿,置于含LumiCycle实时生物发光检测系统的培养箱(37°C,5% CO₂)中连续记录生物发光信号约3-4天。采用Lumicycle分析软件计算生物发光节律的周期长度。
采用Prime Editing 7系统构建PER2 S662G突变D15细胞
使用PE7系统将PER2 S662G点突变引入D15细胞。采用在线工具(http://pegfinder.sidichenlab.org/)设计pegRNA及sgRNA。pegRNA的间隔序列为GCTCGCTGGCACTGCCGGGCA,sgRNA序列为CTTGTCTCCCACATGGACGA。pegRNA的RTT及PBS序列为ACACCCTCTGCCTTGCCCGGCAGTGCCAG。将PE7-EGFP与hML1dn-cherry质粒共转染细胞,1天后经流式细胞分选仪分选EGFP与cherry双阳性D15细胞,于35 mm培养皿中继续培养2天。编辑步骤中,转染3.75 μg PER2 S662G pegRNA及1.25 μg sgRNA质粒,细胞培养3天以完成靶位点编辑,随后进行嘌呤霉素筛选。采用有限稀释法从96孔板中挑选单克隆,通过PCR产物测序确认PER2 S662G点突变细胞。
小鼠品系
CK1δflox小鼠(RRID:IMSR_JAX:010487)由苏州大学徐璎教授惠赠。Mark3敲除小鼠(RRID:IMSR_GPT:T029467)购自Gempharmatech(中国南京)。Tssk2敲除小鼠、Tssk1-Tssk2条件性双敲除小鼠、Mark2条件性敲除小鼠、Mark3条件性敲除小鼠及Mark4条件性敲除小鼠均由CIBR遗传操作中心采用CRISPR/Cas9技术构建(附图S4及S11)。
小鼠节律跑轮活动测定
所有动物实验程序均获CIBR动物护理与使用委员会批准。选取11-24周龄雄性小鼠单笼饲养,自由摄取食物和水。采用低轮廓无线跑轮系统记录节律活动。小鼠在标准光照周期(12小时光照/12小时黑暗)中适应14天,随后转入持续黑暗条件适应21天。每10分钟记录一次跑轮活动数据并绘制活动模式图。采用χ⟡周期图分析法对持续黑暗阶段的跑轮活动记录进行分析,计算节律周期长度。
量化和统计分析
采用GraphPad Prism 8.0(GraphPad Software)进行所有统计分析。详细统计信息均标注于相应图注中。实验结果以平均值±标准误(mean ± SEM)表示,除非另有说明。数据正态性通过Shapiro-Wilk检验进行评估:若数据满足正态分布且方差齐性,则采用参数检验;否则,采用非参数检验(Kruskal-Wallis检验)。具体而言,两组间比较采用非配对双尾Student's t检验;多组与对照组比较采用单因素方差分析(ANOVA)followed by Dunnett's多重比较检验;多组间全面比较采用单因素方差分析followed by Tukey's多重比较检验;不同处理下多组间比较采用双因素方差分析followed by Sidak's多重比较检验。Western blot结果使用ImageJ软件进行灰度值定量,并在GraphPad Prism中绘图;定量数据以均值±标准误表示,每个数据点代表n=3个生物学重复的独立实验。统计显著性标记为:*p < 0.05,**p < 0.01,***p < 0.001,****p < 0.0001。除特殊说明外,n表示生物学独立样本、动物数量或重复实验次数,具体数值详见各图注。所有关键实验均独立重复至少三次,图中展示代表性结果。各实验组样本量(n)的完整汇总信息见表S3和表S4。
附图

图S1. 抗磷酸化S662抗体的特异性验证及CK1激酶磷酸化PER2 S662,与图1相关
(A-C)采用(A)全长hPER2、(B)全长mPER2及(C)hPER2 556-771片段验证抗磷酸化S662抗体的特异性。S662A/S662D(hPER2)或S659A/S659D(mPER2)突变以及λ-磷酸酶处理均可消除抗体识别信号。(D-E)FLAG标签的CK1δ及CK1ε在HEK293T细胞中磷酸化hPER2 556-771片段的S662位点。将CK1δ或CK1ε及其他激酶分别与hPER2 556-771在HEK293T细胞中共表达。转染24小时后收集细胞,进行免疫印迹分析。(F)CK1家族中仅CK1δ和CK1ε可磷酸化全长hPER2的S662位点。将CK1亚家族各成员分别与全长hPER2在HEK293T细胞中共表达。(G)CK1δ及CK1ε对hPER2 S662的剂量依赖性磷酸化。将递增浓度的CK1δ或CK1ε cDNA与hPER2 556-771共转染HEK293T细胞。(H)免疫沉淀的CK1δ及CK1ε在体外直接磷酸化重组PER2(556-771)的S662位点。从HEK293T细胞中分别免疫沉淀CK1亚家族的FLAG标签激酶,检测其对从大肠杆菌纯化的重组hPER2(556-771)的磷酸化能力。

图S2. 重组CK1δ及CK1ε磷酸化PER2 S662,与图1相关
本图所用蛋白均在大肠杆菌中表达并纯化。hPER2底物(rPER2野生型、S662A、S662D)及小鼠PER2底物(r-mPER2野生型、S659A、S659D)均包含556-771位氨基酸残基。重组CK1δ及CK1ε(野生型及激酶失活K38R突变体)均为包含激酶结构域的N端片段(1-317位氨基酸)。其他CK1家族成员(CK1α1、CK1α2、CK1γ1、CK1γ2、CK1γ3)为全长蛋白。蛋白纯度经考马斯亮蓝染色验证,见图S8A、B及D。
(A-B)重组CK1δ野生型磷酸化PER2 S662位点。(C-D)重组CK1ε野生型磷酸化PER2 S662位点。(E-F)重组CK1δ野生型及CK1ε野生型磷酸化小鼠PER2保守位点S659。(G)重组CK1家族成员对PER2 S662磷酸化的特异性。(H-I)重组CK1δ及CK1ε作用于PER2 321-771的米氏常数测定。将约0.2 μg rCK1δ或CK1ε与不同浓度的rPER2 321-771底物于37°C孵育30分钟。免疫印迹结果进行定量分析(n=3个独立生物学重复)。数据以均值±标准误表示。(J)CK1激酶对PER2 S662磷酸化能力的系统发育树总结。"IP"表示从HEK293细胞免疫沉淀的激酶磷酸化PER2 S662的能力,"E. coli"表示在大肠杆菌中表达的激酶磷酸化PER2 S662的能力。

图S3. CK1敲除D15细胞及小鼠中节律的延长,与图1相关
(A-B)采用CRISPR/Cas9技术在D15细胞中敲除CK1δ(A)及CK1ε(B)的策略。(C-D)CK1δ或CK1ε基因敲除延长D15细胞节律周期。采用Lumicycle系统检测CK1δ或CK1ε敲除D15细胞的节律(对照组:n=7;CK1δ敲除组:n=6;CK1ε敲除组:n=6个独立生物学重复)。展示代表性振荡曲线(C)及周期变化统计分析(D)。(E-F)神经元特异性敲除CK1δ小鼠节律周期延长约0.30小时。展示代表性跑轮活动图(F)及持续黑暗条件下跑轮活动周期计算结果(E)。Nestin-Cre;Ck1δ+/+(n=14),Nestin-Cre;Ck1δflox/+(n=9),Nestin-Cre;Ck1δflox/flox(n=11)。(G-H)CK1δ/ε双敲除细胞中存在其他PER2 S662激酶。在亲本及CK1δ/CK1ε双敲除HEK293细胞中表达FLAG-hPER2,检测PER2 S662磷酸化水平。免疫印迹结果采用ImageJ软件进行定量分析(n=3个独立生物学重复)。定量结果显示双敲除克隆中PER2 S662磷酸化仍持续存在(为亲本水平的62%-72%),表明存在其他作用于S662的激酶。统计分析采用单因素方差分析后接Tukey多重比较检验(E);单因素方差分析-Dunnett多重比较检验(D和H);数据以均值±标准误表示。
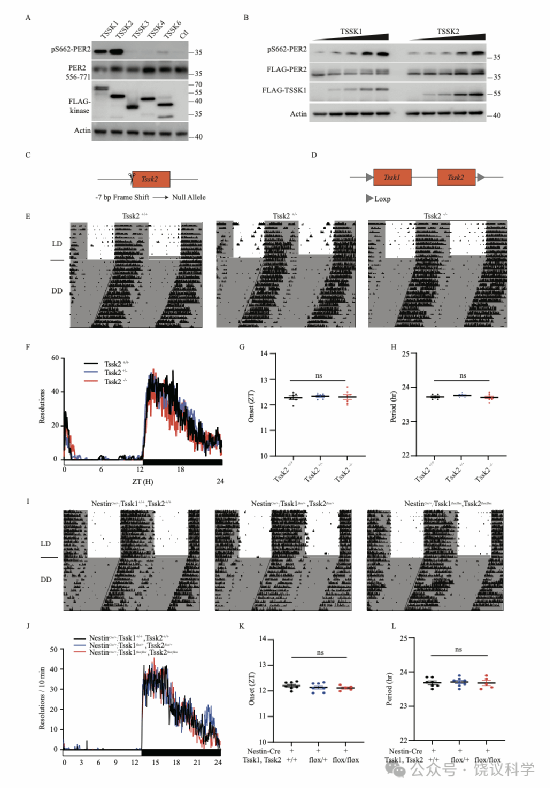
图S4. Tssk1及Tssk2敲除小鼠的节律,与图1相关
(A)TSSK1及TSSK2(而非其他TSSK家族成员)与hPER2(556-771)在HEK293T细胞中共表达时可磷酸化其S662位点。(B)转染TSSK1或TSSK2以剂量依赖性方式增强PER2-S662磷酸化。将递增浓度的TSSK1或TSSK2 cDNA与hPER2 556-771共转染HEK293T细胞。(C)采用CRISPR/Cas9技术构建常规Tssk2敲除小鼠的策略示意图。(D)Tssk1flox/flox-Tssk2flox/flox小鼠的构建策略。(E-L)Tssk2敲除小鼠节律正常。Tssk2+/+(n=6),Tssk2+/-(n=6)及Tssk2-/-(n=7)。展示代表性跑轮活动图(E),光照-黑暗循环条件下每10分钟跑轮转数(F),光照-黑暗循环条件下活动起始时间(G)及持续黑暗条件下周期长度(H)。数据以均值±标准误表示。(I-L)神经元特异性Tssk1-Tssk2双敲除小鼠节律正常。NestinCre;Tssk1+/+Tssk2+/+(n=7),NestinCre;Tssk1+/flox-Tssk2+/flox(n=7)及NestinCre;Tssk1flox/flox-Tssk2flox/flox(n=5)小鼠。展示代表性跑轮活动图(I),光照-黑暗循环条件下每10分钟跑轮转数(J),光照-黑暗循环条件下活动起始时间(K)及持续黑暗条件下周期长度(L)。统计分析采用单因素方差分析-Tukey多重比较检验(G、H、K、L);数据以均值±标准误表示。

图S5. MARK3磷酸化PER2 S662,与图2相关
(A)免疫沉淀的MARK3在体外磷酸化重组PER2 S662。在HEK293细胞中表达FLAG标签的人源MARK3野生型、T211A及T211E突变体,免疫沉淀后与1 μg rPER2 556-771底物进行激酶反应。(B)梯度转染MARK3野生型或T211E突变体增强PER2-S662磷酸化水平。(C-D)从HEK293T细胞免疫沉淀的小鼠MARK2(C)及MARK3(D)在体外磷酸化重组hPER2(556-771)的S662位点。(E-H)敲除HEK293细胞中MARK1(E)、MARK2(F)、MARK3(G)及MARK4(H)基因的策略示意图。(I-J)四个MARK基因同时敲除显著降低PER2 S662磷酸化水平。将FLAG标签的hPER2转染至MARK1-4四基因敲除HEK293细胞中,采用抗磷酸化S662单克隆抗体检测S662磷酸化水平。免疫印迹结果采用ImageJ软件进行定量分析(n=3个独立生物学重复)。与亲本对照细胞相比,三个独立敲除克隆(KO-1、KO-2、KO-3)的磷酸化水平均显著降低60%-67%。统计分析采用单因素方差分析-Dunnett多重比较检验(J);数据以均值±标准误表示。

图S6. 重组MARK3对PER2 S662的直接磷酸化,与图3相关
本图所有实验均使用在大肠杆菌中表达并纯化的重组蛋白进行。采用重组蛋白在体外进行磷酸化测定。(A)PER2 S662可被重组MARK3 T211E活化突变体磷酸化,而野生型MARK3及T211A失活突变体无此活性。(B)MARK3 T211E可磷酸化PER2野生型,但对S662A及S662D突变体无活性。(C)MARK3 T211E作用于PER2的米氏常数测定。将约0.2 μg rMARK3 T208E与不同浓度的rPER2底物于37°C孵育30分钟。免疫印迹结果进行定量分析(n=3个独立生物学重复)并计算Km值。数据以均值±标准误表示。(D)小鼠MARK3 T211E可磷酸化小鼠PER2野生型,但对S659A及S659D突变体无活性。

图S7. 重组ARKs及CK1激酶的表达,与图4相关
(A)在大肠杆菌中表达的ARK构建体总结,注明为全长或片段形式及激活环T到E突变。部分ARKs(如SIK1-3)的全长重组蛋白在大肠杆菌中表达成功,部分未成功。将激活环中保守的苏氨酸残基("Activation Loop Phosphorylation Site"栏所示)突变为谷氨酸(T到E)以构建磷酸模拟突变体。(B)在大肠杆菌中表达的CK1家族蛋白总结。CK1δ及CK1ε(野生型及激酶失活K38R突变体)为包含激酶结构域的N端片段(1-317位氨基酸);其他CK1成员为全长蛋白。因CK1激酶可通过自身磷酸化激活,未在大肠杆菌中表达纯化S到D突变蛋白。

图S8. 在大肠杆菌中表达的重组蛋白,与图4相关
本文所用重组蛋白的考马斯亮蓝染色。取各重组蛋白1 μg,经蛋白上样缓冲液于95°C变性后进行12% SDS-PAGE,考马斯亮蓝R250染色。所有重组激酶(除PER2底物外)均表达为单一融合构建体,带有N端MBP-TEV酶切位点-3xFLAG标签及C端TEV酶切位点-GFP-8xHis标签。TEV酶切位点为烟草蚀纹病毒蛋白酶识别位点。若重组蛋白用作底物,则不进行酶切;若重组蛋白作为激酶进行检测,则先用TEV蛋白酶处理,使激酶部分从融合蛋白中释放。
(A)rCK1α1、rCK1α2、rCK1δ 1-317、rCK1ε 1-317、rCK1γ1、rCK1γ2、rCK1γ3。(B)rPER2 556-771、rPER2 556-771-A、rPER2 556-771-D、rCK1δ 1-317、rCK1δ K38R 1-317、rCK1ε 1-317、rCK1ε K38R 1-317、rPER2 321-771。(C)rMARK2、rMARK2-A、rMARK2-E、rMARK3、rMARK3-A、rMARK3-E。(D)小鼠rPER2 556-771、小鼠rPER2 556-771-A、小鼠rPER2 556-771-D、小鼠rMARK2、小鼠rMARK2-E、小鼠rMARK3、小鼠rMARK3-E、rMAP4K7 1-500。

图S9. 在大肠杆菌中表达的重组蛋白,与图4相关
纯化的ARK家族蛋白考马斯亮蓝染色(每孔1 μg)。(A)rAMPKa1、rAMPKa2、rSIK1、rSIK2、rSIK3、rMARK1、rMARK2、rMARK3、rMARK4、rNUAK1、rNUAK2、rMELK。(B)rAMPKa1-E、rAMPKa2-E、rSIK1-E、rSIK2-E、rSIK3-E、rMARK1-E、rMARK2-E、rMARK3-E、rMARK4-E、rNUAK1-E、rNUAK2-E、rMELK-E。(C)rTSSK1、rTSSK2、rTSSK3、rTSSK4、rTSSK6、rBRSK1、rBRSK2、rHUNK、rNIM1K、rSNRK、rSIK3。(D)rTSSK1-E、rTSSK2-E、rTSSK3-E、rTSSK4-E、rTSSK6-E、rBRSK1-E、rBRSK2-E、rHUNK-E、rNIM1K-E、rSNRK-E、rSIK3-E。

图S10. D15细胞中MARKs的功能研究,与图6相关
(A-D)免疫印迹验证MARKs在D15细胞中的稳定过表达。(E)采用CRISPR-Cas9技术构建MARK单基因敲除D15细胞系(敲除策略见图S6E-S6H)。敲除细胞经PCR验证。(F-H)过表达MARK1或MARK4分别使节律周期延长0.91小时或1.47小时。展示MARK1(F)及MARK4(G)过表达细胞的代表性振荡曲线及周期变化统计分析(H)。对照组:n=7;MARK1过表达组:n=7;MARK4过表达组:n=8个独立生物学重复。(I-K)MARK4敲除缩短节律周期,而MARK1敲除无显著影响。在D15细胞中分别敲除MARK1及MARK4。展示MARK1(I)及MARK4(J)敲除细胞的代表性振荡曲线。MARK4敲除细胞周期长度缩短0.36小时,而MARK1敲除细胞周期无显著变化(K)。对照组:n=7;MARK1敲除组:n=6;MARK4敲除组:n=6个独立生物学重复。(L)PER2 S662G突变D15细胞的构建。经PCR测序确认PER2 S662G点突变细胞。统计分析采用单因素方差分析-Dunnett多重比较检验(H和K);数据以均值±标准误表示。

图S11. 条件性Marks敲除小鼠的构建及Mark4敲除小鼠的节律,与图7相关
(A-C)构建Mark2flox/flox(A)、Mark3flox/flox(B)及Mark4flox/flox(C)小鼠的打靶策略。(D-H)神经元特异性Mark4敲除小鼠节律正常。NestinCre;Mark4+/+(n=5)及NestinCre;Mark4flox/flox(n=6)小鼠。展示代表性跑轮活动图(D和E),光照-黑暗循环条件下每10分钟跑轮转数(F),光照-黑暗循环条件下活动起始时间(G)及持续黑暗条件下周期长度(H)。统计分析采用非配对Student's t检验(G和H);数据以均值±标准误表示。
表S1. 用于筛选hPER2片段磷酸化的288种蛋白激酶cDNA列表,与图1相关


表S2. 寡核苷酸及引物序列,与STAR方法相关


表S3. D15敲除细胞的节律周期,与图6、图S3及图S10相关


表S4. 基因敲除小鼠模型的节律表型,与图7、图S3、图S4及图S11相关

























论文链接
https://doi.org/10.1016/j.chembiol.2026.02.007


